
Introduction
Multiple myeloma is a plasma cell malignancy characterized by the proliferation of monoclonal plasma cells in the bone marrow and excessive production of monoclonal paraprotein (M protein), bone destruction, hypercalcemia, displacement of other hematopoietic cell lines, suppression of bone marrow function and renal damage.1 According to GLOBOCAN estimation, 828 individuals were diagnosed with MM and 475 patients died from the disease in 2020 in Switzerland.2 The 5-year prevalence of multiple myeloma is 2,360, with an incidence of 27.27/100,000 inhabitants. In the period 2013−2017, the 5-year survival rate was 58% for men and 57% for women.3
Although therapies for patients with MM have improved in recent years, the unmet medical need for novel treatments remains high for patients who have been exposed to the main drug classes, including immunomodulatory drugs (IMiDs), proteasome inhibitors (Pis) and CD38-targeting antibodies such as daratumumab and isatuximab, as MM is still an incurable disease.4–6 Data from LocoMMotion, the first prospective study of 92 various real-life standards of care (SoC) regimens in patients with heavily pretreated, triple-class-exposed RRMM showed that there is no clear SoC in this clinical setting and the clinical outcomes with the available therapies are poor.7 Over the past few years, a new generation of immunotherapies has emerged, including drugs directed against BCMA, a novel treatment target for MM due to its highly selective expression in malignant plasma cells.8,9 To date, encouraging results were reported for BCMA-directed CAR T-cell therapies10 like idecabtagene vicleucel11 and ciltacabtagene autoleucel,12 belantamab mafodotin,13 an antibody-drug conjugate (ADC), and bispecific antibodies such as teclistamab and elranatamab.14,15
BCMA-targeted CAR T-cell therapies in RRMM
There are currently two Swissmedic-approved CAR T-cell therapies for clinical use in RRMM, idecabtagene vicleucel and ciltacabtagene autoleucel.16,17 The positive results from pivotal trials on CAR T-cell therapy for MM facilitated the initiation of numerous preclinical and clinical trials on myeloma-directed CAR T-cell products.18
Idecabtagene vicleucel: The first CAR T-cell therapy for triple-class-exposed RRMM
Idecabtagene vicleucel (ide-cel; ABECMA®) was the first CAR T-cell therapy approved for the treatment of adult patients with RRMM who had had ≥3 prior lines of therapy, including an IMiD, a PI and an anti-CD38 antibody, with disease progression on the last therapy.16 This was based on the encouraging results from the pivotal, single-arm, phase II KarMMa trial of ide-cel in this patient population (n=128).19 Updated analysis at a median follow-up of 24.8 months showed that 73% of patients had a response, including 33% with a CR or better (≥CR) and 52% with a very good partial response or better (≥VGPR).20 The overall median progression-free survival (PFS) was 8.6 months and the median overall survival (OS) was 24.8 months, with 12-month and 24-month OS rates of 78% and 51%, respectively. Comparable results were reported across prespecified subgroups, including the number of prior therapies (3 vs ≥4). Ide-cel also showed statistically significant and clinically meaningful improvements in several health-related quality of life (HRQoL) domains and global health status/HRQoL within two months after treatment initiation, with clinically meaningful improvements in fatigue, pain and physical functioning maintained through 15−18 months.21
To understand the long-term experience of patients receiving ide-cel for RRMM, a qualitative study was recently performed on data from post-infusion interviews with 45 patients, which were conducted 6–24 months after ide-cel treatment in the KarMMa trial.22 This longitudinal analysis showed that all patients reported ≥1 ide-cel treatment advantage, mainly related to efficacy (93%), few or no side effects (78%) and avoidance of other treatments (76%). Most patients reported improvements in physical functioning, emotional well-being, social life and future outlook at 6 months post-infusion compared with baseline; these improvements generally remained stable through 18 and 24 months.
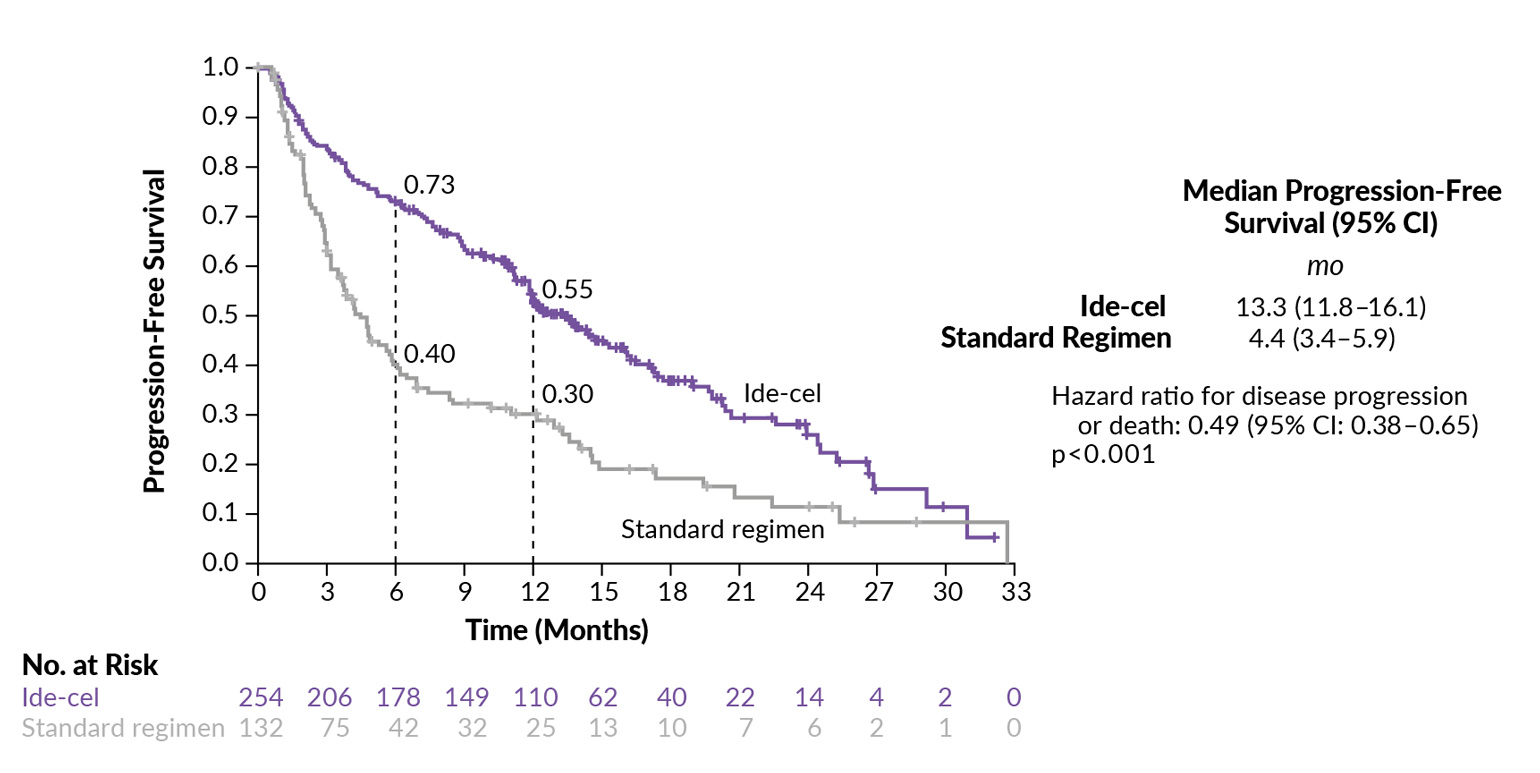
Ide-cel was further assessed versus standard regimens in the open-label, phase III KarMMA-3 trial on adult patients with RRMM who had previously received 2−4 regimens, including an IMiD, PI and daratumumab, and who had disease refractory to the last therapy.23 At baseline, two-thirds of patients had a triple-class-refractory disease and 95% had a daratumumab-refractory disease. At a median follow-up of 18.6 months, the median PFS was significantly prolonged with ide-cel compared with a standard regimen (13.3 months vs 4.4 months; HR: 0.49 [95% CI: 0.38−0.65; p<0.001]) (Figure 1). Ide-cel was also associated with a significantly higher response rate at 71% versus 42% with a standard regimen (p<0.001), including CR rates of 39% and 5%, respectively. Data on OS were immature at the time of this analysis. In terms of safety, grade 3−4 adverse events (AEs) were reported in 93% of patients in the ide-cel arm and 75% of those in the standard-regimen arm. Among patients receiving ide-cel, cytokine release syndrome (CRS) occurred in 88% (grade ≥3: 5%) and investigator-identified neurotoxic effects occurred in 15% (grade ≥3: 3%).
_versus_a_.jpg)
One of the first cohorts of RRMM patients treated with commercial ide-cel was recently reported.24 This real-world study presented experiences with ide-cel in 16 triple-class exposed RRMM patients treated at the University Hospital of Bern Switzerland between June and October 2022. At baseline, the median age was 69 years, 38% of patients had high-risk cytogenetics, 19% Revised International Staging System (R-ISS) stage III and 31% had extramedullary disease; the median number of previous treatment lines was 6. At 3 months, the proportion of patients with overall response was 69%, with a stringent (s)CR rate of 44%, CR rate of 6% and VGPR of 19%. The safety outcomes of ide-cel were consistent with previously published trial data.11 CRS was identified in 94% of patients (grade 1: 88%; grade 2: 6%), immune effector-cell associated neurotoxicity syndrome (ICANS) in 6% of patients (grade 1), febrile neutropenia in 69% and infections in 31% of patients.24 The median time to CRS onset was 0 days. Taken together, these data showed high response rates with a manageable safety profile in patients treated with commercial ide-cel, but the authors noted that prolonged hematologic toxicity remains a major challenge.
Ciltacabtagene autoleucel (cilta-cel): Durable and robust outcomes in RRMM
Ciltacabtagene autoleucel (cilta-cel; CARVYKTI®) is another CAR T-cell therapy indicated for the treatment of adult RRMM patients after three or more prior lines of therapy, including a PI, IMiD and an anti-CD38 antibody.17 This BCMA-directed CAR T-cell therapy with two BCMA binding sites demonstrated unprecedented clinical response and survival outcomes in patients enrolled in the pivotal, phase Ib/II, open-label CARTITUDE-1 trial (n=97).25 The 2-year follow-up analysis showed durable and deep responses with cilta-cel, with nearly all patients achieving an overall response, with an sCR rate of 82.5% and the VGPR rate of 12.4%.26 The ORR benefit was consistent across all prespecified subgroups. The median PFS and the median OS were not reached, with 27-month rates of 54.9% and 70.4% in the overall population, respectively. More than 90% of patients with minimal residual disease (MRD)-evaluable samples (n=61) were MRD negative (threshold 10-5), which was maintained for ≥6 months in 68% and ≥12 months in 55% of patients. Achieving sustained MRD negativity considerably improved PFS and OS rates.
Notably, neurotoxicity signals reported with cilta-cel were different from other CAR T-cell therapies and were discussed in detail by Arber et al. (2022) in the healthbook TIMES Oncology Hematology.10,27 Briefly, 17% (any grade) and 2% (grade 3 or 4) of patients experienced ICANS but a novel type of neurotoxicity was also observed.27 A total of 5% of patients experienced a cluster of cilta-cel-related movement and neurocognitive treatment-related adverse events (MNT). Patients with high tumor burden, CRS grade 2 or higher and any grade of ICANS were more likely to experience MNT-related neurotoxicity. After implementing specific strategies for the prevention and early management of toxicities, the MNT rate dropped below 1% across the cilta-cel program.
Cilta-cel was also evaluated versus SoC, such as pomalidomide plus bortezomib and dexamethasone (PVd) or daratumumab plus pomalidomide and dexamethasone (DPd), in the phase III CARTITUDE-4 trial in patients with relapsed and lenalidomide-refractory MM.28 This study enrolled 419 adult patients who had received 1−3 prior lines of therapy for MM, including an IMiD and PI. At the first pre-specified interim analysis, the study met its primary endpoint of a significant improvement in PFS, which led to the unblinding of the study.29,30 First results showed that the median PFS was not reached in the cilta-cel arm and was 11.8 months in the SoC arm, with a 12-month PFS rate of 76% and 49%, respectively (HR: 0.26 [95% CI: 0.18−0.38]; p<0.0001) (Figure 2). This PFS was maintained across all key subgroups, including the number of prior lines of therapy (1 vs 2−3). The ORR was 84.6% and 67.3% with cilta-cel and SoC, including an sCR/CR rate of 73.1% and 21.8%, respectively. The 12-month duration of response (DoR) was 84.7% in the cilta-cel arm and 63.0% in the SoC arm, with a median DoR of not reached and 16.6 months, respectively. MRD negativity was reported in 60.6% of patients receiving cilta-cel and 15.6% of those receiving SoC (odds ratio [OR]: 8.7; p<0.0001). OS data were immature but showed a trend favoring cilta-cel (HR: 0.78 [95% CI: 0.5−0.12]; p=0.26). In terms of safety, no new signals were reported. CRS, mainly of grade 1−2, occurred in 76.1% of patients who received cilta-cel; there were no grade 4 or 5 events. ICANS occurred in 4.5% of patients and all events were grade 1−2. These data place cilta-cel as a potential SoC in patients with relapsed and lenalidomide-refractory MM.
_in_cartitude-4.jpg)
The effectiveness of cilta-cel versus real-world clinical practice (RWCP) was assessed in inverse probability weighting (IPW)-adjusted comparisons using individual patient data from CARTITUDE-1 (enrolled, n=113; infused, n=97) and LocoMMotion (enrolled, n=248; included, n=170), a prospective, multinational study of patients with triple-class-exposed RRMM.31 In LocoMMotion, the most frequent regimes were carfilzomib plus dexamethasone (13.7%), pomalidomide plus cyclophosphamide and dexamethasone (13.3%) and pomalidomide plus dexamethasone (11.3%). Results showed that cilta-cel consistently yielded superior response rates and survival outcomes compared with RWCP. More specifically, patients treated with cilta-cel were 3.12-fold more likely to respond to treatment versus RWCP (ORR: 97.9% versus 42.9%; p<0.0001). Rates of ≥VGPR and ≥CR were 94.8% and 82.5% with cilta-cel, compared with 17.6% and 0.6% with RWCP. The median PFS and OS in the cilta-cel group were not reached, while they were 4.34 months (HR: 0.15 [95% CI: 0.08−0.29]; p<0.0001) and 11.33 months (HR: 0.20 [95% CI: 0.09−0.41]; p<0.0001), respectively, in the adjusted RWCP population. The analysis of patient-reported outcomes data further demonstrated that patients receiving cilta-cel experienced incremental improvements in quality of life over time with regards to EuroQol visual analog scales (EQ VAS) and global health status (GHS), while improvements with RWCP were considerably smaller. In terms of safety, any grade (100% vs 83.5%) and grade 3−4 (93.8% vs 49.2%) AEs were more common in patients treated with cilta-cel versus RWCP patients; however, the overall safety profile was manageable. An unadjusted comparison of all AEs also indicated higher AE rates for cilta-cel compared with RWCP across organ classes.
Similar results were recently reported from the multicenter, retrospective Belgium Comparator study in Multiple Myeloma (BELCOMM) exploring longitudinal data of patient characteristics and outcomes across treatment lines from triple-class-exposed RRMM patients receiving RWCP who fulfilled the main inclusion criteria from CARTITUDE-1.32 By using the RWCP cohort as an external control arm for CARTITUDE-1, this analysis showed that after inverse probability weighting (IPW)-average treatment effect in the treated population (ATT) re-weighting, the ORR was 96.9% in cilta-cel-treated patients and 27.0% in RWCP-treated patients (RR: 3.00 [95% CI: 2.32−3.89]; p<0.0001), including ≥VGPR rates of 93.8% and 8.0%, respectively (RR: 11.51 [95% CI: 6.38−20.77]; p<0.0001). Furthermore, the medians for PFS, OS and time to next treatment (TTNT) were not reached in the cilta-cel cohort and were significantly shorter in the ATT-reweighted RWCP cohort. Notably, most patients in the RWCP group were heavily pretreated and refractory to several therapies.
BCMA-directed antibodies
Belantamab mafodotin: ADC therapy for RRMM
Belantamab mafodotin33 is a first-in-class, highly selective antibody-drug conjugate (ADC) that consists of an anti-BCMA monoclonal antibody conjugated to monomethyl auristatin F, a microtubule-disrupting agent. Upon binding to BCMA on plasma cells, belantamab mafodotin is internalized into the cell, where monomethyl auristatin F (MMAF) induces apoptosis. The efficacy and safety of belantamab mafodotin were demonstrated in the open-label, phase II DREAMM-2 study on RRMM patients who received ≥4 prior therapies, including a PI, an IMiD and an anti-CD38 antibody.34,35 In this study, patients received single-agent belantamab mafodotin at either 2.5 mg/kg (n=97) or 3.4 mg/kg (n=99) intravenously in 3-week cycles until disease progression or unacceptable toxicity. In the final analysis of this trial, belantamab mafodotin yielded rapid, deep, durable and clinically meaningful responses in this patient population.36 In the 2.5 mg/kg and 3.4 mg/kg groups, the ORR was 32% and 35%, respectively, including ≥VGPR rates of 19% and 24%. The median OS was 15.3 months in the 2.5 mg/kg cohort and 14.0 months in the 3.4 mg/kg cohort. Among patients who achieved ≥VGPR, the median OS ranged between 30.7 months (2.5 mg/kg group) and 35.5 months (3.4 mg/kg group). Similarly, patients with ≥VGPR had considerably longer PFS (14.0 months and 16.8 months) compared with the overall populations (2.8 months and 3.9 months). Belantamab mafodotin was well-tolerated with maintained quality of life despite the increased frequency of ocular events, which were manageable with dose modifications and supportive measures. Based on positive results from DREAMM-2, belantamab mafodotin was approved in June 2022 by the Swissmedic as monotherapy for the treatment of adult RRMM patients who have received ≥4 prior lines of therapy and whose disease is refractory to at ≥1 PI, one IMiD and one anti-CD38 monoclonal antibody and who have had disease progression on the last therapy.33
More recently, data were reported from the open-label, phase III DREAMM-3 trial, which investigated single-agent belantamab mafodotin versus pomalidomide plus low-dose dexamethasone in 325 RRMM patients.37 The study failed to meet its primary endpoint of PFS, with a median PFS of 11.2 months in the belantamab mafodotin arm and 7.0 months in the pomalidomide arm (HR: 1.03 [95% CI: 0.72−1.47]). The ORR was 41% with belantamab mafodotin versus 36% with pomalidomide, however considerably more patients receiving belantamab mafodotin achieved ≥VGPR (25% vs 8%). With 37.5% of maturity of OS data, the median OS was 21.2 months in the belantamab mafodotin arm versus 21.1 months in the pomalidomide arm (HR: 1.14 [95% CI: 0.77−1.68]). Following these results, the EMA recommended non-renewal of authorization of belantamab mafodotin38 which is also not approved in Switzerland anymore. Belantamab mafodotin is currently being evaluated in combination with bortezomib and dexamethasone in the phase III DREAMM-7 trial and in combination with pomalidomide and dexamethasone in the phase III DREAMM-8 trial.
The antitumor activity of belantamab mafodotin was corroborated in an observational, retrospective, multicenter study in 106 adult patients with heavily pretreated RRMM.39 Approximately half of the patients were triple-class refractory and 11.3% were penta-class refractory. Approximately 38.0% of patients had an overall response, with a median DoR of 9 months. The median OS was 9.3 months and the median PFS was 3.5 months. Treatment was delayed in 52.9% of patients, including 36.5% due to treatment-related toxicity. In the safety population (n=104), the most common toxicity were ocular AEs, mainly grade ≤2, present in nearly half of the patients. Overall, these data are consistent with the results from DREAMM-2 in terms of efficacy and safety.
Teclistamab: First BiTE therapy for RRMM
Teclistamab is an off-the-shelf, bispecific IgG4 antibody that binds both BCMA and CD3 to redirect CD3+ T cells to BCMA+ myeloma cells, prompting T-cell activation followed by tumor cell death and lysis.14 It is approved for the treatment of adult RRMM patients who have previously received ≥3 lines of therapy, including a PI, an IMiD and an anti-CD38 antibody.40 In the registrational phase I/II MajesTEC-1 study (n=165), 63.0% of patients treated with teclistamab had a response and 39.4% achieved ≥CR.41 At a median follow-up of 14.1 months, the median PFS was 11.3 months, which compared favorably to other agents in the RRMM setting. Neurotoxic events occurred in 14.5% of patients, including ICANS (3.0% of patients; all grade 1 or 2). A detailed analysis of safety data additionally showed that most cases of CRS occurred during the step-up dosing schedule and were mainly grade 1 (50.3%) or grade 2 (21.2%); one patient experienced grade 3 CRS.42 All CRS cases resolved and none led to treatment discontinuation. One-third of patients had at least one CRS event; CRS recurrence was reduced in patients treated with tocilizumab for the first CRS event compared with patients who were not treated with it (20.0% vs 62.2%). Teclistamab is currently being evaluated in combination with daratumumab in the phase III MajesTEC-3 trial.43
Due to the lack of head-to-head trials, two recent studies evaluated the comparative efficacy of teclistamab versus currently available treatments in patients with triple-class-exposed RRMM patients.44,45 Using indirect, adjusted treatment comparisons, Mateos et al. (2023) reported significantly improved outcomes with teclistamab versus physician’s choice of therapy (PC).44 In detail, this study included individual patient-level data from patients treated with teclistamab in MajesTEC-1, while an external control arm was created from patients in long-term follow-up of four clinical trials of daratumumab (APOLLO, POLLUX, CASTOR and EQUULEUS) who were treated with PC after discontinuation of trial treatments. In the primary analysis, teclistamab versus PC was associated with significantly improved clinical outcomes after adjustment. These included ORR (OR: 4.81), ≥VGPR rate (OR: 12.07), OS (HR: 0.54), PFS (HR: 0.59) and TTNT (HR: 0.32) (all p≤0.0001). The results of the fully adjusted model, which considered all 14 prognostic factors, were consistent with the primary analysis.
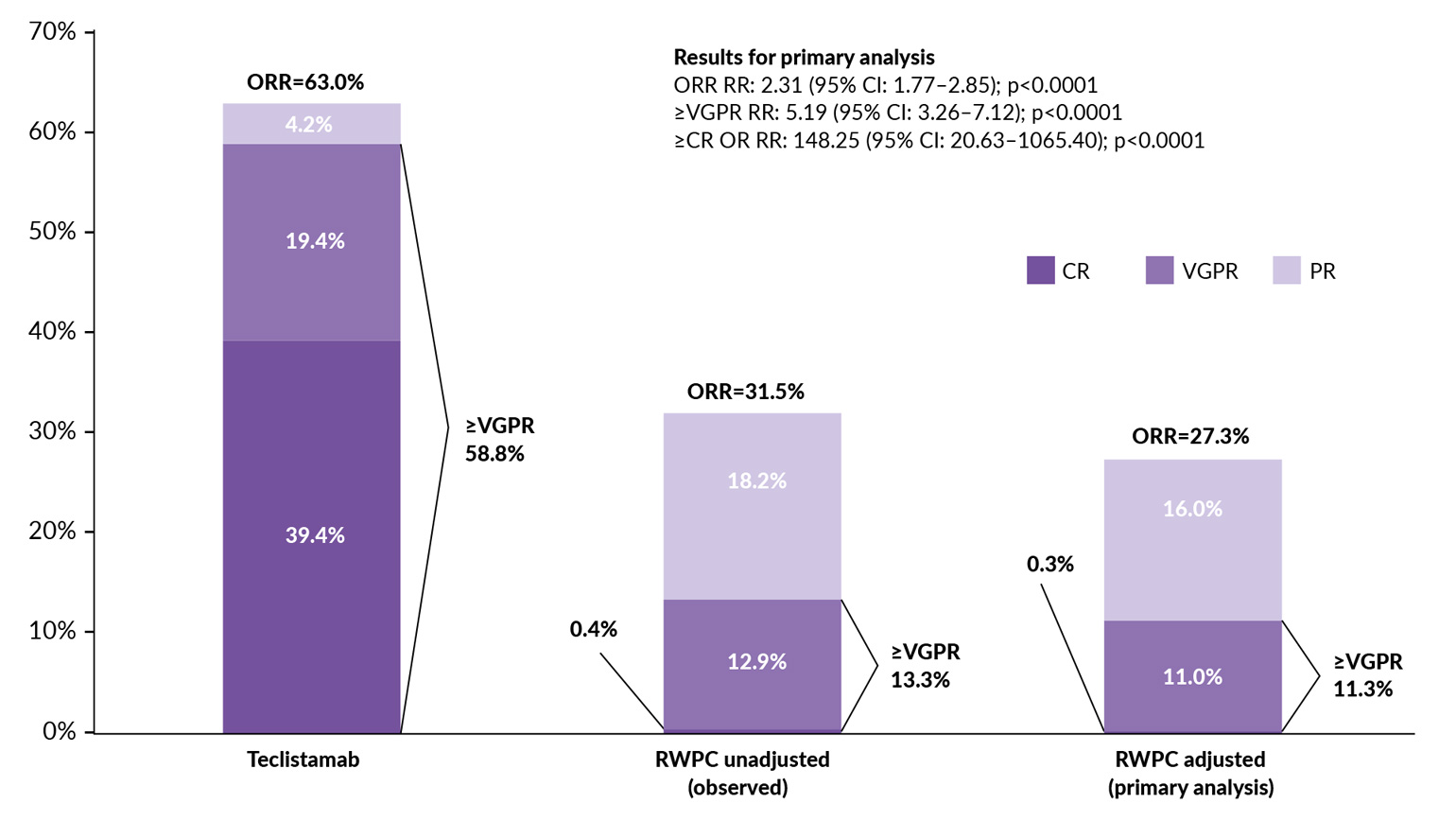
To assess the relative effectiveness of teclistamab versus real-world physician’s choice of therapy (RWPC), another study performed adjusted comparisons using individual patient data from MajesTEC-1 and LocoMMotion.45 After IPW-ATT adjustment, teclistamab demonstrated significantly improved response rates versus RWPC, including ORR (response rate ratio [RR]: 2.31), ≥VGPR (RR: 5.19) and ≥CR (RR: 148.25) (all p<0.0001) (Figure 3). In adjustment comparisons, teclistamab versus RWPC also achieved improved DoR (median, 18.43 months vs 5.78 months; HR: 0.32 [95% CI: 0.19–0.54]; p<0.0001) and PFS (median, 11.30 months vs 4.14 months; HR: 0.48 [95% CI: 0.35–0.65]; p<0.0001). OS was numerically prolonged with teclistamab versus RWPC (HR: 0.77 [95% CI: 0.55–1.09]; p=0.1419). Altogether, these data showed improved effectiveness of teclistamab versus RWPC, highlighting its clinical benefit as a novel and effective treatment for patients with triple-class-exposed RRMM.44,45
.jpg)
Teclistamab was further investigated in combination with talquetamab, a first-in-class GPRC5DxCD3 bispecific antibody, for the treatment of patients with RRMM. The first results from the phase Ib RedirectTT-1 study showed that this dual bispecific regimen yielded promising responses with a clinically manageable safety profile in RRMM patients, including those with extramedullary disease.46 In the overall population (n=93), the ORR was 86.6% after a median follow-up of 13.4 months, including a CR+sCR rate of 40.2% and a VGPR rate of 36.6%. The median time to first response was 1.97 months and the median DoR was not estimable. In patients receiving the recommended phase II regimen (RP2R) (n=34), the ORR was 96.3%, with a CR+sCR rate of 40.7% and a VGPR rate of 48.1%, at a median follow-up of 8.1 months. The median time to first response was 1.48 months and the median DoR was not estimable. Among those with extramedullary disease (all soft tissue plasmacytomas), the ORR across all dose levels (n=35) was 71.4% (CR+sCR: 21.2%; VGPR: 28.6%) and 85.7% among patients receiving the RP2R (n=11) (CR+sCR: 28.6%; VGPR: 42.9%). The median DoR was 12.9 months and not estimable, respectively. Based on these encouraging results, RedirectTT-1 expansion cohort in extramedullary disease (EMD) has been planned.
Elranatamab: New option for heavily pretreated RRMM
In September 2023, another BCMAxCD3 bispecific antibody, elranatamab, received temporary authorization by Swissmedic for the treatment of adult patients with RRMM refractory to at least one IMiD, one PI and one anti-CD38 monoclonal antibody, and who have shown progression to the last therapy.47 This approval was based on data from the ongoing single-arm, phase II MagnetisMM-3 trial demonstrating that elranatamab induced deep and durable responses in this patient population (n=123).15 The primary endpoint of confirmed ORR was 61.0%, including a ≥CR rate of 35.0% and a ≥VGPR of 56.1%. At a median follow-up of 14.7 months, the median DoR, PFS and OS (secondary endpoints) have not been reached, with a 15-month DoR rate of 71.5%, PFS rate of 50.9% and OS rate of 56.7%. Expectedly, the safety profile reported with elranatamab was comparable to that reported with teclistamab as the two agents have a similar mechanism of action.15,41 However, the incidence of CRS and ICANS was slightly lower with elranatamab at 56.3% among patients who received premedication and a two step-up priming dose regimen (n=119) (57.7% in the total population [n=123]) and 3.4% (vs 72.1% [grade 3: 0.6%] and 3.0% with teclistamab), although most of these cases were of low grade with both drugs.
Conclusion
-
Therapies targeting BCMA will play an important role in MM treatments, but further studies are needed to clarify their impact on clinical practice.
-
BCMA-directed CAR T-cell therapies have demonstrated favorable efficacy and safety in patients with RRMM, with currently two approved constructs in Switzerland (ide-cel and cilta-cel).16,17 Based on these encouraging results, both therapies are being evaluated for use in earlier lines of therapy.23,48
-
Belantamab mafodotin is currently being investigated in combination with other anti-myeloma treatments.49,50
-
Teclistamab and elranatamab are the first BCMA-targeted BiTEs that represent effective therapies for heavily pretreated RRMM patients with limited treatment options.15,41
Conflict of interest
Adrian Schmidt received consultancy honoraria and travel grants from Novartis, Takeda, Incyte, Sanofi, Celgene, Amgen and Janssen. These funding entities did not play a role in the development of the manuscript and did not influence its content in any way.
Funding
The author has declared that no financial support was received from any organization for the submitted work.
Author contributions
The author created and approved the final manuscript.