Introduction
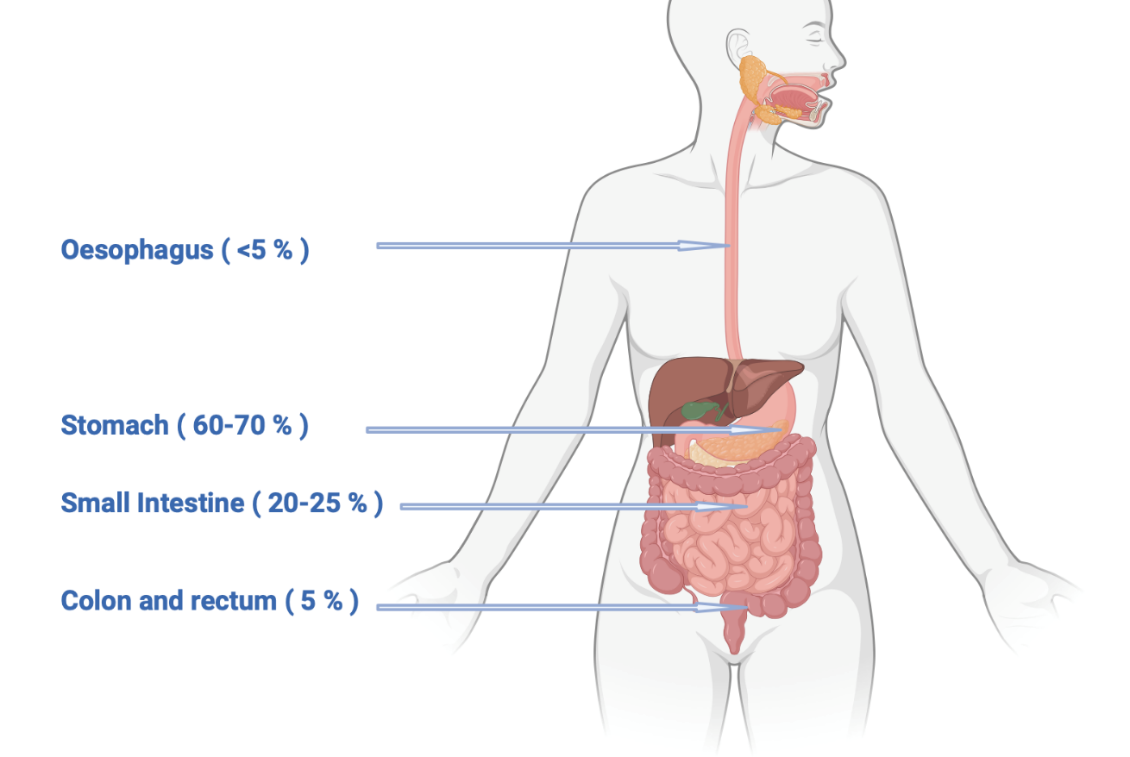
Gastrointestinal stromal tumors (GISTs) make up 20% of sarcomatous tumors and 1−2% of primary gastrointestinal cancers. The median age at presentation is 60−65 years, with the stomach being the most common location (60−70%), followed by the small intestine (20−25%), colon and rectum (5%), and esophagus (<5%) (Figure 1).1 GISTs can also affect children and young adults, particularly in the case of genetic predispositions, for example, primary familial GIST syndrome, and neurofibromatosis type 1.2 GISTs often do not cause symptoms and are usually discovered incidentally during radiology examination. The most common symptoms associated with GISTs are abdominal pain or gastrointestinal bleeding. Endoscopic and radiological findings can vary, with small tumors appearing as homogeneous lesions, while larger tumors may exhibit heterogeneous enhancement, irregular margins, central necrosis, and signs of hemorrhage.3,4

GISTs originate from Cajal’s interstitial cells, or their precursors, and consistently express the KIT protein/receptor. Histologically, GISTs can be classified into two main types: spindle-cell GISTs, primarily associated with mutated KIT or BRAF genes (occurring in 70% of cases), and epithelioid-cell GISTs (20%), predominantly linked to platelet-derived growth factor receptor A (PDGFRA) or succinate dehydrogenase (SDH) gene mutations. Approximately 10% of GISTs display mixed morphology.5,6 Immunostaining for DOG-1 (discovered on GIST-1) has high sensitivity and specificity and is present in 88% of GISTs.7 KIT staining is detected in 95% of GISTs and is highly sensitive but not specific. In PDGFRA-mutated GISTs, the sensitivity of these markers decreases to 9% and 79%, respectively.8
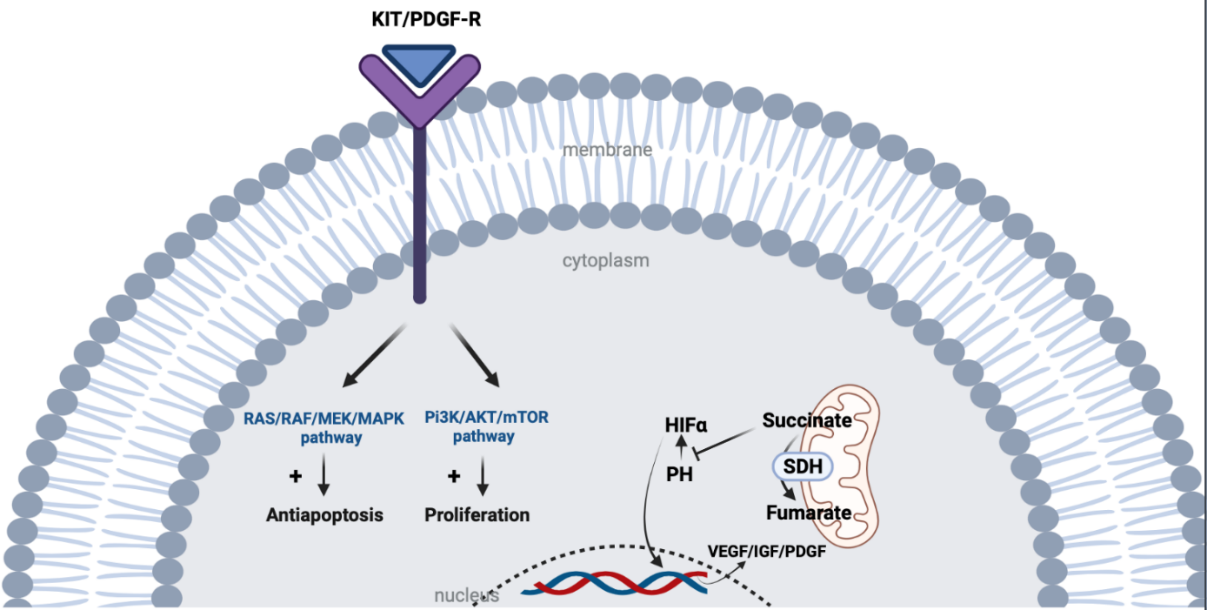
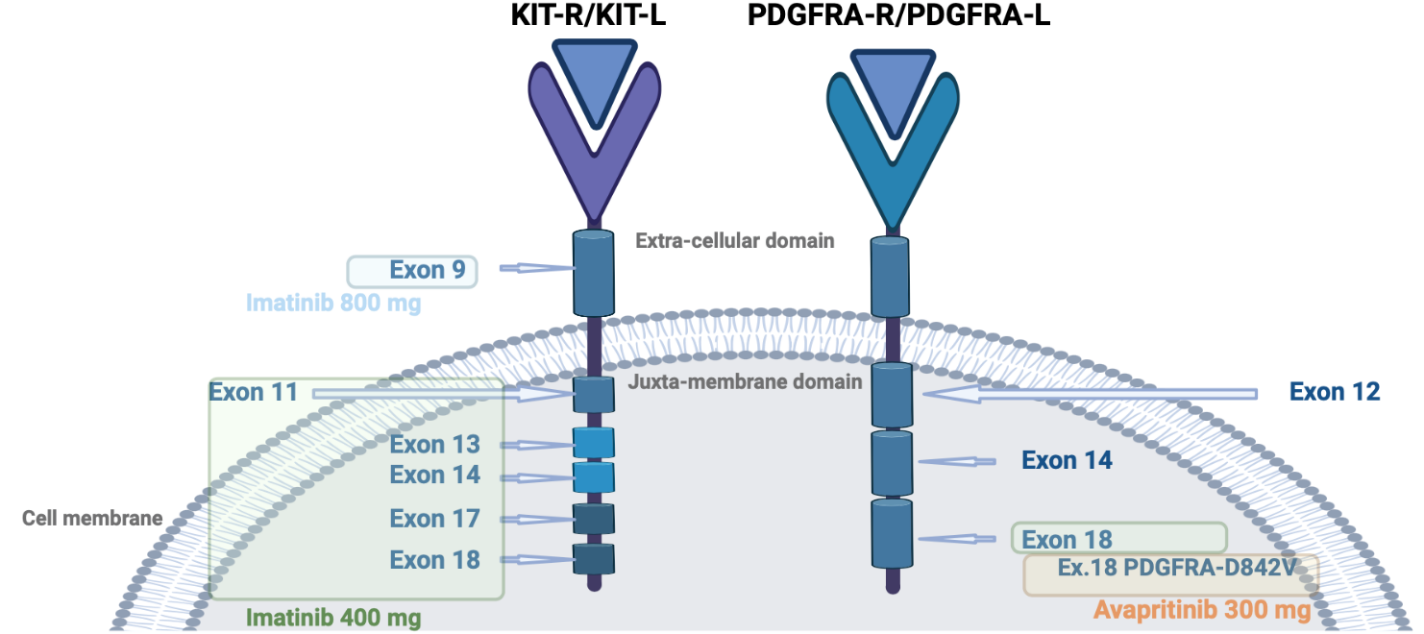
KIT is a proto-oncogene that encodes the KIT tyrosine kinase (TK) receptor. It consists of two main regions: the receptor regulatory domain (extracellular region, transmembrane region, and juxta-membrane domain) and the cytoplasmic region containing the TK domain (including TK1 and TK2 domains). The activation loop (encoded by exon 17) on the TK2 domain stabilizes the active state of the KIT receptor. The binding of the ligand to KIT leads to the activation of tyrosine kinase activity and the initiation of downstream pathways, including RAS/RAF/MAPK and PI3K/Akt/mTOR, promoting cellular proliferation and inhibiting apoptosis. Activating mutations in KIT result in the constitutive ligand-independent activation of the receptor. PDGFRA is structurally similar to KIT (Figure 2 and Figure 3).6
-deficient_gastro.png)
The KIT gene contains the majority of gain-of-function mutations in GISTs, with the most frequent mutations occurring in exon 11.6 Exon 9 mutations are commonly found in the bowel and account for around 10% of GISTs.9 Various types of genetic alterations, such as deletions, point mutations, duplications, insertions, and inversions, have been identified in the KIT gene. Exon 11 deletions, particularly codons 557 and 558, are associated with a poorer prognosis compared to exon 11-point mutations. Mutations in exons 13, 17, and 18 are rare.10
Around 5 to 10% of GISTs mutations are in PDGFRA, particularly in exons 12, 14, and 18. PDGFRA mutations are more commonly found in the stomach, with exon 18 being the most frequently mutated region. The D842V mutation in exon 18 accounts for 70% of PDGFRA-mutant cases, while exons 12 or 14 are rarely mutated. KIT and PDGFRA mutations are mutually exclusive (Figure 3).11

Although extensively sequenced, 10−15% of GISTs are classified as wild-type. Contemporary techniques such as whole genome sequencing have enabled the detection of infrequent KIT or PDGFRA mutations in some of these instances. Additionally, certain cases have been linked to NTRK and FGFR1 fusion events, modifications in the RAS-MAPK pathway due to BRAF mutations, NF1 mutations, or SDHA deficiency caused by a germline mutation in the SDH complex or SDHC promoter methylation. SDH deficiency causes the accumulation of succinate that blocks the degradation of HIF1 by prolyl hydroxylase (PH) resulting in the expression of growth factors (Figure 2).
In the case of metastatic wild-type GISTs, it is essential to refer the patient to a reference center for appropriate molecular analysis as some rare genetic alterations (e.g. NTRK fusion) can lead to treatment options.12
Localized GISTs
Surgery is considered the primary treatment modality for operable GISTs, as long as no major functional losses are anticipated. It is crucial to achieve complete resection of the tumor and avoid tumor rupture.3 For patients at high risk of recurrence, post-operative imatinib is recommended for a minimum of 3 years, provided it is well tolerated, as it has proven benefit on overall survival (OS) (5-year OS rate: 92.0% vs 81.7%; HR: 0.45 [95% CI: 0.22−0.89]; p=0.02), but the optimal duration of adjuvant imatinib has not been definitively established, and ongoing clinical trials are investigating different durations, to determine the most effective treatment approach.13
If R0 surgery is not possible, or function-sparing surgery is considered, or if surgical resection could be safer after cytoreduction by reducing the risk of bleeding and tumor rupture, pre-treatment with imatinib is the standard approach, provided the tumor’s mutation profile is sensitive. However, a limitation could be the difficulty in reliably evaluating the mitotic count for accurate risk stratification during biopsy, which can make decisions regarding post-operative therapy more challenging.3 Another limitation is the lack of tumor markers to monitor tumor response.
To determine the risk of recurrence, different scoring systems are used:
-
The Miettinen classification, also known as the Armed Forces Institute of Pathology (AFIP) classification, takes into account three parameters: the mitotic index, tumor size, and tumor location along the gastrointestinal (GI) tract.
-
Joensuu’s classification, also referred to as the modified National Cancer Institute (NIH) classification, incorporates the aforementioned parameters while also considering the negative impact of tumor perforation. It aims to better differentiate GISTs between intermediate and high risk.14
All these classifications categorize patients into very low, low, intermediate, and high-risk groups based on their likelihood of recurrence. The decision for adjuvant treatment depends on the risk score and the mutational status of the tumor.15 For instance, patients with the PDGFRA D842V mutation do not receive adjuvant treatment since this mutation confers resistance to imatinib.16
A TNM classification is available (UICC TNM8) but is not yet widely used in practice.3
Intermediate-risk tumors remain difficult to characterize, as they are a heterogeneous group for which the decision on adjuvant treatment must be made on a case-by-case basis and after multidisciplinary discussion. Molecular prognostic factors, such as the level of tumor genome rearrangement, are currently under investigation and being evaluated specifically in intermediate-risk GISTs as part of the GI-GIST trial (NCT02576080).17
Management of metastatic GISTs
First-line treatment
Imatinib
In 2002, imatinib, a KIT, PDGFRA, and BCR-ABL tyrosine kinase inhibitor, was approved after a phase II multicenter, randomized study that evaluated the safety and efficacy of imatinib in 147 patients with advanced GIST.18 A subsequent phase III trial, the EORTC 62005 study19 evaluated its effectiveness at daily dosages of 400 mg and 800 mg. The trial showed a benefit in progression-free survival (PFS), but no OS benefit at the higher dosage with higher toxicity. However, patients with GISTs carrying exon 9 mutations derived a significant benefit from the 800 mg dosage. As a result, the standard dose for advanced GIST patients with KIT exon 9 mutations receiving first-line imatinib is 800 mg.
Oligometastatic disease
Retrospective studies on cytoreductive surgery for recurrent or metastatic GISTs have shown no benefits for advanced or recurrent cases. The primary treatment approach remains systemic therapy with imatinib. While cytoreductive surgery may be considered for stable or responsive disease, and in cases of limited progression on imatinib with feasible resection, careful case selection is necessary. Prospective studies are lacking to further investigate the effectiveness of cytoreductive surgery in this context.20
Duration of imatinib in the metastatic setting
Imatinib is currently given until disease progression or intolerance in metastatic cases. The ongoing study, GIST-TEN (NCT05009927) explores the potential of discontinuing imatinib after 10 years of treatment in a highly selected patient group compared to continuing the treatment.21 However, long-term responders to imatinib are rare, and most patients experience disease progression within 2 to 3 years of starting treatment.
Primary resistance to treatment
Pseudo-resistance
Drug interactions between TKIs and other medications like proton pump inhibitors can be a major source of underdosing and hence lack of response to imatinib.22 Inter-patient variability in imatinib blood levels can be influenced by genetic polymorphisms of cytochrome P450.23 In case of disease progression, increasing the dose to 800 mg daily can overcome some resistance.24
Compliance issues and constitutional low dosage can be significant factors to consider when suspecting pseudo-progression. Monitoring imatinib dosage can help achieve effective plasma concentrations as an observational study showed that only 33.3% of patients achieved durable effective imatinib concentrations.25,26 Demetri et al. (2009) conducted a study on a small group of patients, demonstrating that imatinib plasma levels above 1,100 ng/mL were associated with clinical benefits and a longer time to disease progression.27 In a study involving imatinib-treated patients diagnosed with chronic myeloid leukemia or GIST, 30% of patients interrupted therapy for at least 30 consecutive days within the first year.28
PDGFRA Exon 18 D842V mutation
This mutation makes up 75% of PDGFRA mutations and confers primary resistance to imatinib.29 Avapritinib was specifically developed to target this mutation, and the NAVIGATOR study demonstrated its high efficacy with an overall response rate (ORR) above 90% and a duration of response of 70% at 1 year.30 However, cognitive dysfunction was observed in 37.0% of patients treated with 300 mg of avapritinib. In the trial, toxicity was manageable with close monitoring of adverse events and dose modifications generally allowed patients to continue treatment.
Non-KIT/PDGFRA molecular subtypes
Wild-type GISTs are typically non-responsive to imatinib. Succinate dehydrogenase (SDH)-deficient GISTs account for 5 to 8% of GISTs,31 and approximately 20−40% of wildtype-GISTs show defects in the SDH complex, which can be caused by germline mutations. Limited data are available for SDH-deficient GISTs due to their rarity, but sunitinib demonstrated better response rates than imatinib in a retrospective analysis of 87 patients with SDH-deficient GISTs.32 Trials with sunitinib and regorafenib have shown efficacy, and a novel third-generation TKI, olverembatinib, is being investigated.33,34 Promising treatments for this subgroup include specific HIF1a inhibitors (NCT04895748,35 NCT0492407536) and temozolomide (NCT0355638437).
Tumor agnostic alterations like NTRK fusions and BRAF V600E mutations make up a small part of this subgroup and can respond respectively to TRK inhibitors like entrectinib or larotrectinib38 or BRAF inhibitors.39
A small percentage of wildtype GISTs have NF1 mutations. Approximately 7% of individuals with NF1 develop GISTs.31 Currently, there is no established treatment for NF1-mutated GISTs, as they do not respond to TKIs. Surgery is the primary option for these patients. However, these tumors tend to be less aggressive.40
Secondary resistance
In the second line, 67% of patients have one or more secondary mutations, including KIT exon 17 (encodes activation loop), or exon 13 and 14 (that encodes the drug/ATP binding pocket of the receptor) mutations.41 Mechanisms of secondary resistance to avapritinib in PDGFRA-mutant GIST have been recently described and involve compound mutations of exons 13, 14, and 15 of PDGFRA that show cross-resistance to all other drugs that inhibit PDGFRA.42
Second-line treatment
Sunitinib was approved in 2006 for advanced or metastatic GIST patients who had disease progression or intolerance to imatinib.43 It targets KIT/PDGFRA and has anti-angiogenic activity by inhibiting VEGFR. A placebo-controlled phase III trial demonstrated a longer time to progression (27.3 weeks vs 6.4 weeks, p<0.0001) in patients treated with sunitinib versus placebo, and a higher response rate in KIT exon 9 mutated GIST, while it had poor activity against exon 17 and 18 mutated GISTs.
Ripretinib was compared to sunitinib in the phase III INTRIGUE trial as a second-line treatment after imatinib failure.44 Although ripretinib did not meet the primary endpoint of superiority, it showed meaningful clinical activity, fewer grade 3/4 adverse events, and improved tolerability. In the exon 11 subgroup, PFS was not statistically different between ripretinib and sunitinib, but ripretinib had a higher ORR. OS data are not mature. Sunitinib showed improved PFS in the exon 9 subgroup.
Third-line treatment options
Regorafenib, an anti-angiogenic multi-kinase inhibitor targeting VEGFR, demonstrated significant improvement in median PFS in a phase III study.45 In the study, 240 patients with advanced GISTs were randomized to receive regorafenib or placebo, with a median PFS of 4.8 months versus 0.9 months, respectively (HR: 0.27, p<0.0001). Regorafenib is approved as a third-line treatment following imatinib and sunitinib. Unlike sunitinib, regorafenib remains effective against exon 17 mutations, making it a preferred option for patients with primary or secondary mutations in exon 17.
Pazopanib, tested as a third-line treatment after imatinib and sunitinib failure, demonstrated modest benefit. The combination of pazopanib with best supportive care resulted in a median PFS of 3.4 months, compared to 2.3 months with best supportive care alone (HR: 0.59 [95% CI: 0.37−0.96]; p=0.03).46
In the VOYAGER trial, a phase III randomized study, avapritinib was compared to regorafenib as a third-line treatment.47 The primary endpoint of improved PFS was not met. There was no significant difference in median PFS between avapritinib and regorafenib for patients with molecularly unselected, advanced GISTs.
Despite previous resistance to imatinib, rechallenge is possible and has shown improved PFS versus placebo in cases where disease progressed after the failure of imatinib and sunitinib. This suggests that residual disease still contains clones that remain sensitive to imatinib.48
Fourth-line therapy
Ripretinib, a potent TKI targeting KIT, PDGFRA, PDGFRB, TIE2, VEGFR2, and BRAF, was approved based on the INVICTUS trial.49 This double-blind, placebo-controlled trial included 129 participants with advanced GISTs who had progression after imatinib, sunitinib, and regorafenib, with crossover allowed. The trial demonstrated a significant improvement in both PFS and OS in the ripretinib arm compared to the placebo arm (PFS: 6.3 months vs 1 month, HR: 0.15, p<0.0001; OS: 15.1 months vs 6.6 months, HR: 0.36, p=0.0004).
Doubling the dose from 150 mg per day to 2x150 mg resulted in a 3.7-month increase in PFS for patients who experienced progression on the lower dose. This higher dosage can be considered an option when facing progression on the initial treatment.50
Pimitespib, an inhibitor of heat shock protein 90 (HSP90) that stabilizes KIT and PDGFRA, demonstrated improved PFS and OS in the CHAPTERGIST-301 trial.51 This randomized phase III study included previously treated advanced GIST patients. The therapy has been approved as a fourth-line treatment for metastatic GISTs in Japan. However, patients receiving pimitespib reported visual impairments, such as night blindness, retinal vein occlusion, and visual impairment, which resolved upon discontinuation.
Future perspectives
In a randomized phase II trial with 40 patients, the effectiveness of anti–programmed death-1 (PD-1)/PD-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) medications, both alone and in combination, was explored in advanced GISTs. The trial revealed only modest response rates for nivolumab, with or without ipilimumab.52 Other combinations, such as ipilimumab with dasatinib, epacadostat with pembrolizumab, and pembrolizumab with metronomic cyclophosphamide, demonstrated minimal clinical efficacy with a low ORR. Ongoing trials are investigating different combinations, such as avelumab with axitinib or regorafenib, spartalizumab with imatinib or TNO155/ribociclib, a CDK4/6 inhibitor.53 Further evaluation of immunotherapy in GISTs through prospective trials is needed, potentially guided by new biomarkers.
In addition, lenvatinib, a broad-spectrum TKI, is being investigated in the phase II LENVAGIST study (NCT04193553).54 Bezuclastinib (CGT9486), a KIT D816V inhibitor, has shown clinical benefits in an early phase trial and is currently being tested with or without sunitinib in a phase III clinical trial (NCT05208047).55 Another pan KIT inhibitor called THE-630 is undergoing testing in a phase I/II study and has shown promising preclinical results (NCT05160168).56
Finally, as a further step towards precision oncology, the feasibility of molecular monitoring via liquid biopsy is the subject of numerous studies and appears to be a promising approach. Data concerning sensitivity in relation to tumor volume are not yet well elucidated (49).
Conclusion
In spite of the rarity of GISTs, multicentric clinical trials have led to the approval of four lines of treatment. With the progress in molecular diagnostics and systematic sequencing, new targets and treatments are likely to come to light. The emergence of resistance due to secondary mutations caused by TKIs remains a significant challenge. Tailoring treatments through repeated biopsies and non-invasive liquid biopsies at progression could be promising approaches to improve and personalize treatments. Finally, there is much research ongoing with new broad-spectrum KIT TKIs and combination approaches, all aiming to prevent or overcome known emergence resistance mechanisms. To conclude, GISTs are an early success for precision oncology, offering patients a tailored therapeutic approach to a rare disease.
Conflicts of Interest
Thibaud Koessler received fees for consulting or advisory roles from Merck Sharp & Dohme (MSD), Bristol-Myers Squibb (BMS), Lilly, Roche, Boehringer Ingelheim and Servier. Alex Friedlaender received consulting fees from Amgen, AstraZeneca, Roche, Astellas, Takeda, Bristol-Myers Squibb, Merck Sharpe Dohme, Pfizer, Merck, Novartis and Janssen. These funding entities did not play a role in the development of the manuscript and did not influence its content in any way. Other authors have declared that the manuscript was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research received no external funding.
Author Contributions
All authors contributed to and approved the final manuscript.