Introduction
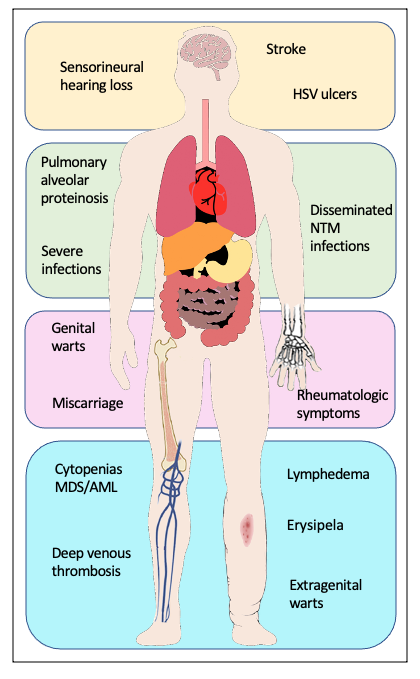
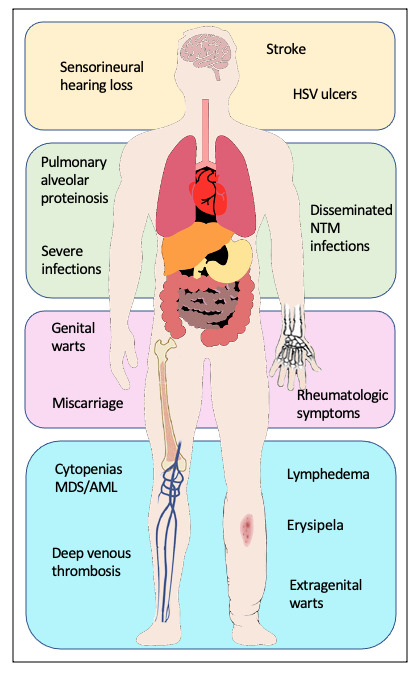
Genetic predisposition to myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) has been an emerging topic over recent years. Mutations in the GATA2 gene were found to be the common genetic cause of four syndromes: Emberger syndrome,1 mycobacterial infections (MonoMAC) syndrome,2 dendritic cell/monocytopenia/natural killer (NK)-cell/B-cell lymphoid deficiency (DCML)3 and familial MDS/AML.4 Additionally, GATA2 deficiency has been linked to aplastic anemia5 and chronic neutropenia.6 Patients may present with manifestations that overlap the clinically described syndromes2,7,8 and are thus all considered part of a single autosomal dominant genetic disorder with varying presentations.7 The clinical presentation of individuals with germline GATA2 mutations is heterogeneous. Some present without any hematopoietic or organ system manifestations prior to the development of MDS or AML,4,7,9 while others may have features of the syndromic presentations (Figure 1):
-
Emberger syndrome, first described in 1979,10 classically features primary lymphedema, sensorineural deafness, cutaneous warts and a predisposition for MDS/AML. A low CD4/CD8 T-cell ratio is also associated with the disease.1
-
MonoMAC syndrome is characterized by profound monocytopenia, B-cell and NK-cell deficiency, resulting in immunodeficiency, and infection with Mycobacterium avium complex, a predisposition for MDS/AML.11 In addition, it is associated with susceptibility to (disseminated) infection with other nontuberculous mycobacteria (NTM), viral and fungal infections, and pulmonary alveolar proteinosis. Several autoimmune phenomena and other rheumatologic symptoms have been described.12,13
-
DCML deficiency is similar to MonoMAC syndrome but with additional dendritic cell (DC) deficiency.13
For patients presenting with MDS at a young age of <45–5014,15 years without a history of chemotherapy or radiation therapy and/or who have a family history suggestive of a familial predisposition (including nonhematological manifestations), testing for GATA2 mutation should be considered. When a diagnosis of GATA2 deficiency is made, a multidisciplinary care team, including hematology, infectious disease, pulmonary and vascular specialists, is needed to manage the multiple affected organ systems. Currently, the only therapeutic option with curative potential is allogeneic hematopoietic stem cell transplantation (alloHSCT). Taking into account the mostly familial occurrence of the disease, often an unrelated matched donor search will be performed. The optimal time frame for alloHSCT has not yet been defined. Management of other medical parameters (vaccinations, chemoprophylaxis, antibiotics, stockings, lymphatic drainage etc.) is of great importance.
Methods
Initially, the two index patients (father and son) had a full medical examination, including personal and family history, blood parameters and genetic analysis, bone marrow (BM) cytogenetic analysis and next-generation sequencing (NGS). In the second step, the other members of the pedigree were investigated over three generations. Medical reports were available from the deceased mother/grandmother of the two index patients. All patients described in this report provided their written informed consent.
Genetic testing for GATA2 mutations in the two index patients was performed at the University Hospital Bern. DNA was extracted from leukocytes from the peripheral blood, using the NucleoSpin tissue kit (Macherey-Nagel). Using the primers described by Pasquet et al. (2013),6 the gene was amplified by polymerase chain reaction (PCR) for Sanger sequencing. With the availability of NGS at the University Hospital Zurich, further testing, including all close relatives, was carried out there, using the TruSight Myeloid Sequencing Panel (Illumina) on the MiSeq (Illumina) platform.
A literature search on GATA2 deficiency was carried out in PubMed using the terms “GATA2”, “GATA2 deficiency” and “Emberger syndrome”. Included were English articles published until April 2020.
Case Report
Two patients (father [Patient 2] and son [Patient 4]) were referred to our service by their general practitioner who remarked unilateral lymphedema and fluctuating cytopenias in both.
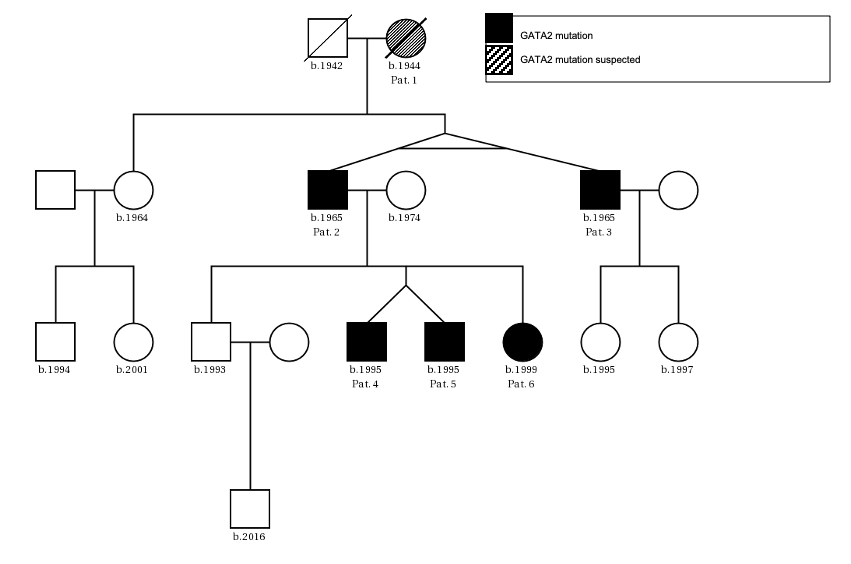
The patients’ family history was noteworthy for the deceased mother of Patient 2 (referred to as Patient 1) who had been diagnosed with MDS at age 47. Otherwise, there were no known hematological diseases in the family. A thorough investigation of the pedigree of the two index patients then led us to a total of six newly diagnosed patients with GATA2 deficiency with the phenotype of Emberger syndrome (Figure 2). A summary of the investigated family members is reported in Table 1.
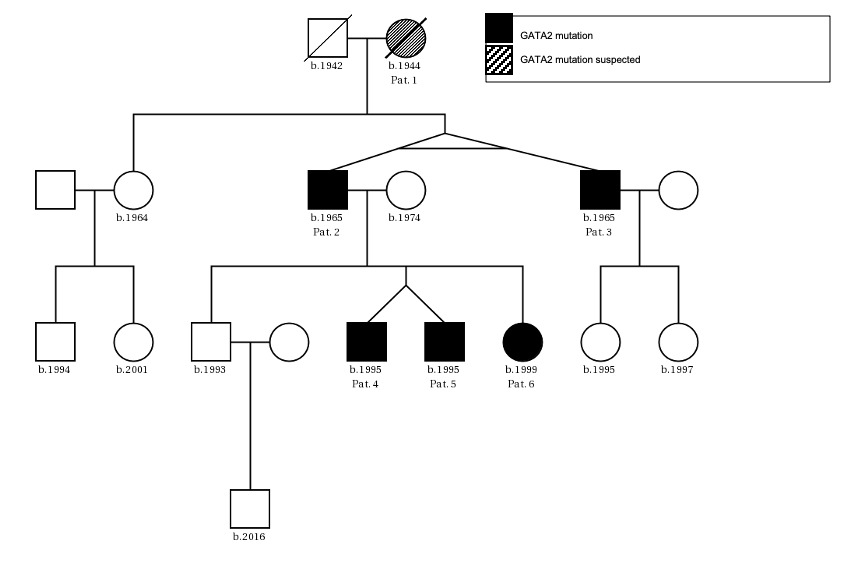
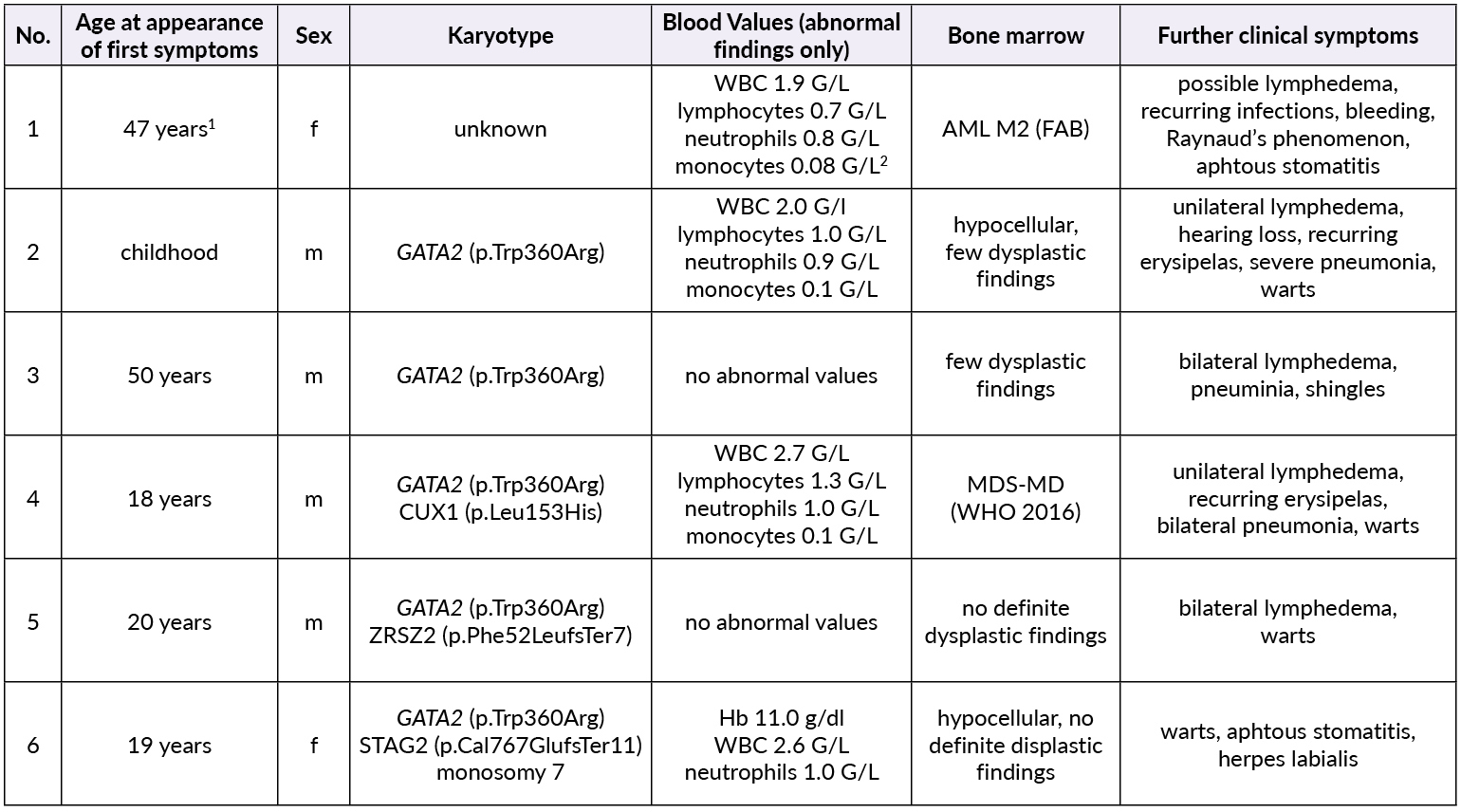
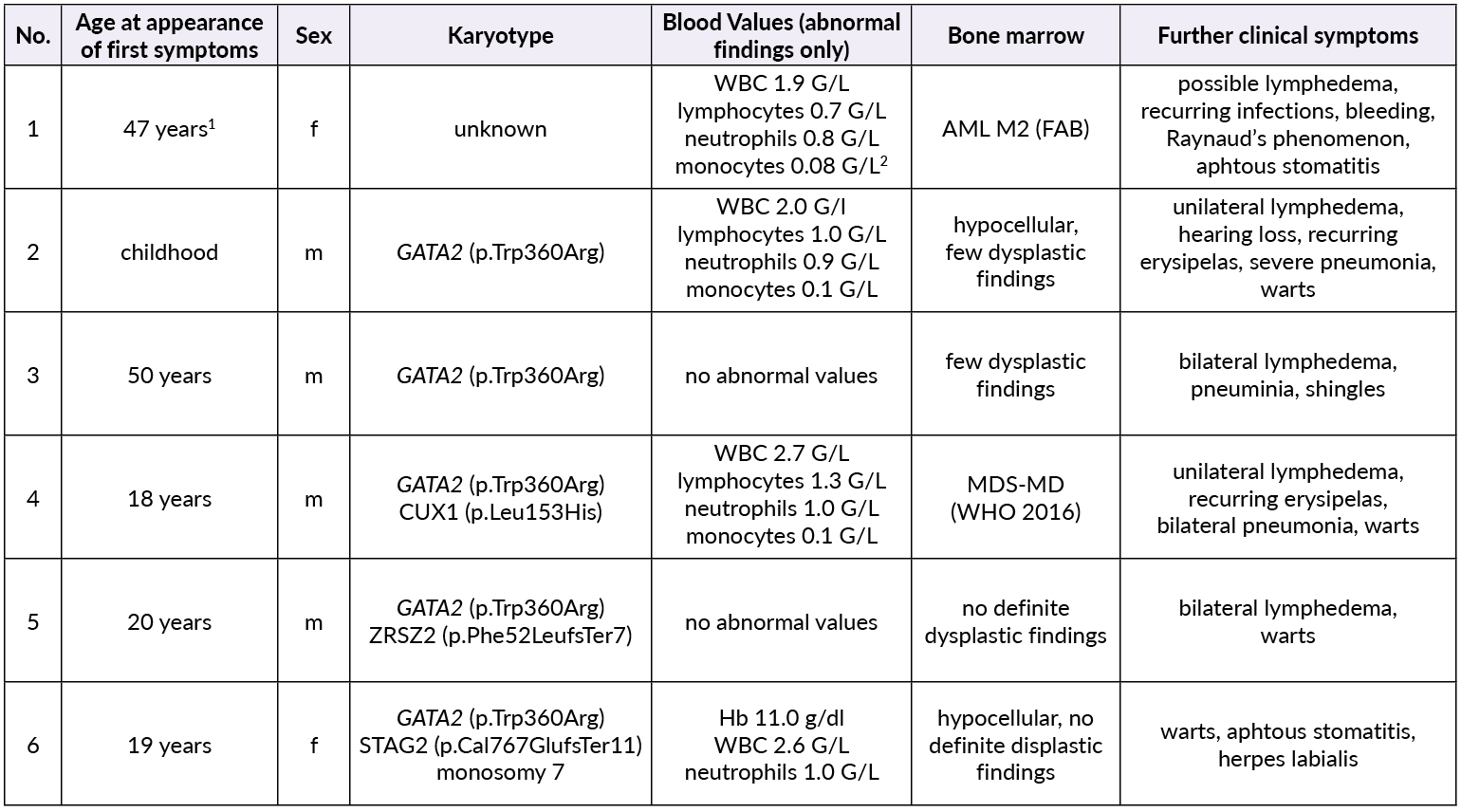
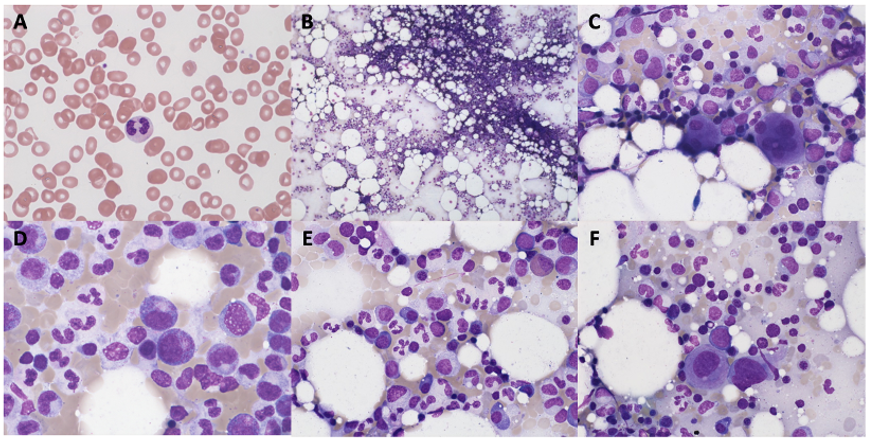
Patient 1 (female, born 1944) had been diagnosed with hypoplastic MDS at 47 years of age following pleuropneumonia. Diagnostic work-up showed a low-risk situation and due to the sufficient peripheral blood values (neutrophils 1.3 G/L, platelets 159 G/L, no anemia), a “watch and wait” approach was chosen. Ten years later, the patient progressed to AML with multiple infectious complications. Allogenic transplantation was performed with rapid relapse five months later. Patient 1 died soon thereafter at the age of 59 years. No GATA2 analysis was performed.
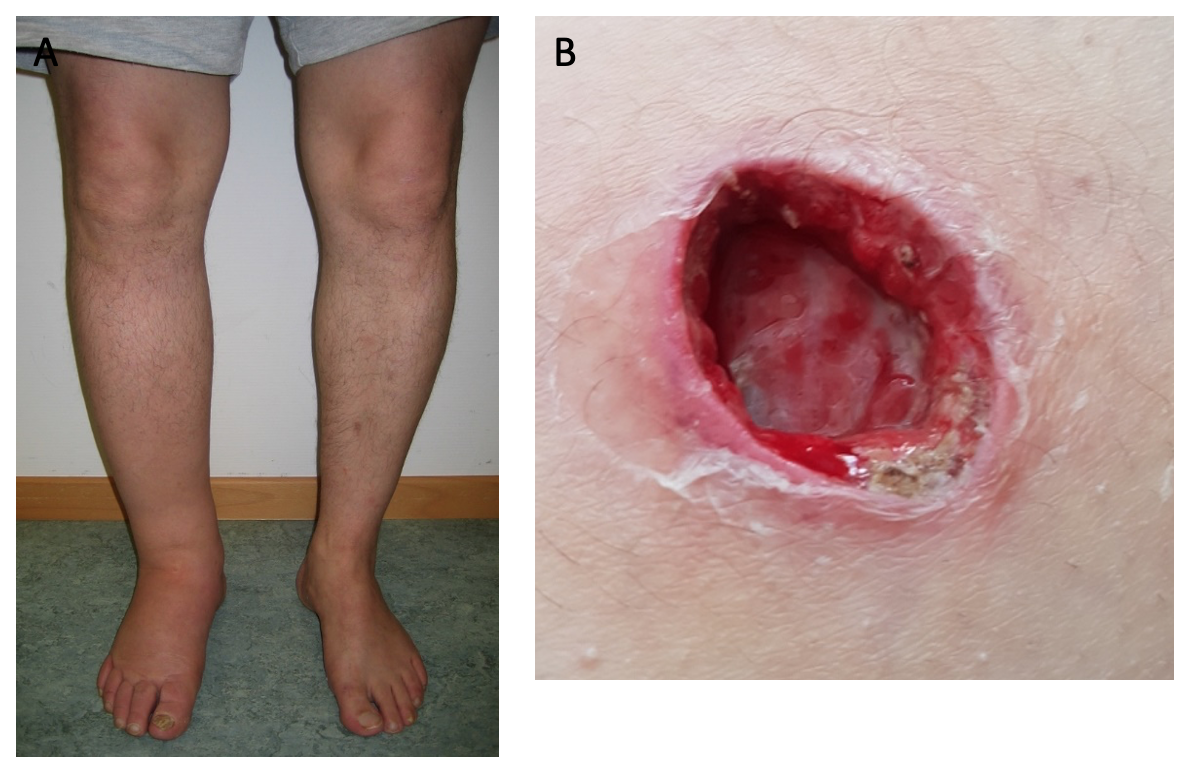
Patient 2 (male, born 1965; index patient) presented with congenital lymphedema of the right leg (Figure 3A), an abundant history of about 25 erysipelas (many resulting in the need for antibiotic treatment and hospitalization) and mild, fluctuating neutropenia (0.9 G/L). Personal history revealed further complications like recurring plantar warts, fungal infections of toes and nails, several events of pneumonia, moderate sensorineural hearing loss and ptosis of the left eye. BM biopsy was hypocellular and showed some dysplastic features, though not enough to be conclusive of MDS. Genotyping revealed a normal karyotype. Experimental genetic testing revealed a GATA2 mutation W360R, affecting the second zinc finger region of the protein.
_patient_2_and_b)_patient_4.png)
Patient 3 (male, born 1965) is a monozygotic twin of Patient 2. Bilateral lymphedema occurred much later than in his brother, at about 50 years of age. Peripheral blood values were normal, the personal history revealed severe pneumonia and a bout of shingles. He also reported impaired hearing but no other known complications of Emberger syndrome. A baseline BM examination showed similar findings to those of his twin, including a normal karyotype and GATA2 mutation.
Patient 4 (male, born 1995; index patient) became conspicuous in his youth with fluctuating lymphocytopenia and thrombocytopenia. At the age of 18 years, lymphedema of the left leg occurred together with repeated erysipelas, some of them requiring hospitalization. Further complications were severe bilateral pneumonia, a history of warts and a vast cutaneous abscess on the left thigh measuring 10 cm (Figure 3B) in diameter. Borderline hypogammaglobulinemia was identified. BM examination revealed a normocellular and sporadically hypocellular BM, with findings consistent with MDS with multilineage dysplasia (MDS-MLD) (World Health Organization [WHO] 2016) (Figure 4). Genotyping revealed a normal karyotype and experimental genetic testing the same GATA2 mutation. NGS further revealed a CUX1 mutation of unknown significance, which has not yet been described (Table 1).
_blood_smear_and_bf)_bone_marrow_biopsy_(bm)_of_patient_4.png)
Patient 5 (male, born 1995) is a dizygotic twin of Patient 4. He became symptomatic at the age of 20 years with bilateral lymphedema of the legs. His personal history included extensive warts on both hands and feet, but only one severe infection following an open fracture of his left arm in childhood. Blood values were normal and BM had no signs of dysplasia. GATA2 mutation was confirmed by NGS, where a truncating mutation in ZRSR2 was found (Table 1). He is the only one with a 10/10 human leukocyte antigens (HLA) matching to the unaffected brother.
Patient 6 (female, born 1999) is the younger sister of patients 4 and 5. She had a severe unspecified infection at the age of one, with no available records. Like her deceased grandmother (Patient 1), she suffers from recurrent stomatitis. She also reported the appearance of small blisters on her face if subjected to cold temperatures. Mild anemia and neutropenia (hemoglobin 110 G/L, neutrophils 0.9 G/L) were present. Further examinations showed a hypocellular BM without definitive dysplastic features or blasts, but with a presence of monosomy 7, GATA2 mutation and a truncating mutation in STAG2 (Table 1).
Patients 2−6 are being followed closely by the local hematologist for blood values, further genetic aberrations and phenotype. All were vaccinated against human papillomavirus (HPV) and annually against influenza. Lymphedema was treated extensively with physical measures (tapes, lymphatic drainage) while warts were handled topically.
Review of the literature
Genetics
GATA2 is located on the long arm of chromosome 3 at position 21.3 (3q21.3).16 It encodes for a zinc finger transcription factor that is a member of a highly conserved family of transcription factors binding to the consensus DNA sequence (T/A)GATA(A/G) and thus regulates multiple target genes.17 GATA2 is involved in hematopoiesis18 (essential for the development and maintenance of the pool of hematopoietic stem cells [HSC] and progenitor cells19), urogenital development,20 neural development21,22 and genesis of lymph vessels.23,24 Additionally, it is found to be highly involved in megakaryocyte development.25 Mutations in GATA2 can be largely divided into 3 groups: 1) truncating nonsense mutations, frameshifts and large gene deletions before or within zinc finger 2 (ZF2*)* domain; 2) missense mutations and in-frame deletions in ZF2; 3) non-coding mutations in intron 5 within the regulatory site.1–4,23,26,27 The genetic mutation found in our cohort (p.W360R) is a missense mutation in ZF2, which has been previously reported by Zhang et al. (2015).28 It disrupts a highly conserved LWRR motif, which can be found in both zinc fingers across the GATA family and is predicted to disturb the folding of ZF2,28 thus interfering with the DNA binding of GATA2. GATA2 mutation is usually transmitted by autosomal dominant inheritance (germline mutation), but sporadic occurrence is possible.29 Somatic mutations in GATA2 are rare and sometimes encountered in MDS30 or AML.31 One particular gain of function mutation (p.L359V) is associated with blast transformation of chronic myeloid leukemia,32 suggesting that regulation of GATA2 within a narrow range is critical.7 A clear genotype-phenotype relationship has not been established so far. Nevertheless, some phenotypic components appear to be dependent on the type of mutation. For instance, frameshift mutations are associated with earlier disease manifestation and more severe immune deficits, while carrying a lower risk of malignancy as compared with missense mutations.33,34
Epidemiology
The prevalence of GATA2 deficiency is unknown. In a small series of familial MDS/AML cases, GATA2 mutations were identified in 4 of 13 concerned families (33%).4 Respectively, in a study with idiopathic BM failure, GATA2 mutations were identified in 5 of 71 patients, being the most commonly identified in an 85-gene panel.28 In an international study of pediatric and adolescent cases, germline GATA2 mutations were identified in 7% of patients with primary MDS and none with secondary MDS.9 Notably, only half of these patients with identified GATA2 mutation showed symptoms indicative of Emberger syndrome, MonoMAC or DCML. Among patients with monosomy 7, the prevalence reached 37%. Yet, the absence of a suggestive family history should not rule out GATA2 deficiency, because germline mutations in GATA2 are known to occur de novo.2,9,28 In a series of 57 patients with germline GATA2 mutations, analyzed by the National Institute of Health (NIH), the age at the initial clinical presentation was 20 years (range: 5 months to 78 years).7 Initial clinical manifestations were infections (64%; 32% viral, 28% disseminated mycobacterial, 4% invasive), MDS/AML (21%) and lymphedema (9%). Notably, 50% of 20-year-old and 16% of 40-year-old patients remained asymptomatic. Clinical manifestations affected overall survival with only 67% surviving 20 years after the onset of initial symptoms.7,8
Clinical findings
As GATA2 is involved in the development of different tissues, mutations in GATA2 can lead to urogenital (malformation, miscarriage), endocrine (hypothyroidism), vascular (thromboembolism, lymphedema), rheumatologic (arthralgia, panniculitis, erythema nodosum) and constitutional changes (dysmorphic features such as long tapering fingers, neck webbing, epicanthic folds, and bi- and unilateral ptosis), as well as autoimmune diseases (lupus, primary biliary cirrhosis and multiple sclerosis). The most important findings are summarized in Figure 1.
The most commonly found clinical features are (severe or recurrent) infections, particularly viral infections with HPV (persistent warts, dysplasia), herpes virus (severe primary infection, recurrent stomatitis) and varicella-zoster virus (VZV) (severe cases).7,35 Moreover, (disseminated) non-tuberculous mycobacteria (NTM) and other bacterial infections (skin and soft tissue infections, pneumonia, sepsis), as well as invasive fungal infections, are not uncommon.7,35
Blood and bone marrow
Hematologic parameters may be normal before the development of MDS4,7 or may show monocytopenia, lymphopenias of B, NK and CD4+ T cells,7 and/or, less frequently, chronic neutropenia.6 Cytopenias may progress over time in asymptomatic individuals. Overall, MDS/AML develops in approximately 50−70% of affected individuals.33,36 BM aspirations and biopsies of patients with GATA2 deficiency often reveal significant BM fibrosis (70−75% vs 5−10% in de novo MDS) and a hypocellular BM, whereas hypercellular BM may be an early sign of progression.11,37 Since GATA2 is also highly involved in megakaryocyte development,25 megakaryocytes may give morphological clues towards GATA2 deficiency: both atypically large and small megakaryocytes (micromegakaryocytes) are possible, with peripheralized and separated nuclear lobes.38 Karyotype is frequently aberrant, most commonly with monosomy 7 or trisomy 8.4,7,9,39,40 Children with MDS and monosomy 7 should further be investigated for GATA2 mutation.9 Furthermore, somatic mutations in ASXL1 are frequently present at the time of malignant transformation.41 As in sporadic disease, ASXL1 mutation is associated with poor prognosis.41,42
Diagnostic work-up
If GATA2 mutation is suspected (Figure 1), a baseline peripheral blood draw, as well as a BM cytology and biopsy (with karyotype analysis), should be performed; nowadays, NGS is mandatory.14 A careful inspection of the skin (warts, abscesses, ptosis, etc.) followed by pulmonary exploration is advised. Family and personal history are of inestimable value. Regular monitoring of blood values with differentials is highly recommended to detect any changes suggestive of both (worsening) immunodeficiency or development of MDS/AML. If any worrisome changes are seen, a repeat of the BM examination should be considered.
Therapy
Initially, therapy is often symptomatic containing antibiotics, virostatics, lymphatic drainage, tapes, etc. Patients should be closely monitored for any sign of malignancy or immunodeficiency. The only therapeutic action with a curative approach is alloHSCT but data on its timing remain vague and sparse. AlloHSCT is recommended earlier than in de novo MDS,43 especially in carriers of ASXL1 mutations,44 depending on frequency of transfusions, clonal cytogenetic abnormalities, grading of dysplasia or severity of recurrent infections, among other factors.14 To date, there are only two small prospective studies on alloHSCT in patients with GATA2 mutations.37,45 A larger prospective trial is currently ongoing.46 Larger datasets exist in a retrospective manner, notably a pediatric cohort with 57 patients,9 as well as a mixed-age cohort with 79 patients.34 Both studies were not primarily focused on the therapeutic approach but provided some data, which otherwise is largely anecdotal. Nevertheless, experts generally agree that alloHSCT should be performed, especially as it is the only curative option for both MDS/AML and many organ manifestations.
Prophylaxis
Due to the susceptibility to viral infections, vaccination against HPV and VZV, as well as the yearly influenza vaccination, should be considered.7 Otherwise, viral infections could pose a life-threatening risk with worsening immunodeficiency.
Discussion
Emberger syndrome is, among others, a syndromic presentation of GATA2 deficiency, which presents with lymphedema, warts, deafness and propensity to (often life-threatening) infections, as well as the development of MDS/AML.1–3,7,40 Our series of five patients with confirmed and one patient with highly suspected GATA2 haploinsufficiency over three generations illustrates some key points in the management of GATA2 deficiency syndromes. First, suspicion of the presence of an underlying disorder is indispensable. Here, the general practitioner noticed uncommon concordant features in the father and son (both patients of his) and referred them to the local hematologist. Second, the evolution of MDS at a young age should always raise suspicion of an underlying genetic disposition.15 If confirmed, familial testing should be initiated. Third, although all carriers of the same GATA2 mutation, clinical course may be rather different within the same family, denying genotype-phenotype correlation to a certain extent. Nevertheless, our cohort consistently presented with mainly Emberger-linked symptoms. Fourth, once a diagnosis is established, regular clinical and laboratory check-ups every three to six months (depending mainly on blood values, the presence of further prognostically relevant aberrations and mutations, as well as associated syndromes) are crucial to monitor the disease and its complications. Fifth, due to the scarcity of relevant studies, it is difficult to define the optimal time point to proceed to alloHSCT, which is the only curative approach. Most experts propose not to wait as long as in MDS without genetic disposition.14,43 In our case, there was one unaffected sibling in each generation of the pedigree, with Patient 5 being the only one with a related possible donor. Therefore, an unrelated matched donor search should be initiated early. Alternatively, haploidentical-related donors may frequently be available, however better strategies to prevent fatal graft versus host disease (GvHD) and graft rejection are needed.47
Conflicts of interest/Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Funding
No funds, grants or other support was received.
Author Contributions
All authors contributed to and approved the final manuscript.
Ethics approval
For this case report, an ethical approval is not necessary, since it does not fall under the Swiss Human Research Act. Informed consent for the usage of patient data has been obtained.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
The participants have consented to the submission of the case report to the journal.