

Introduction
Anemia is the most common blood cytopenia that is frequently found in elderly patients and is associated with increased morbidity and mortality.1 Overall, the prevalence of anemia increases sharply in the general population after the age of 50, reaching >20% in individuals aged 85 years or older.2 After ruling out common causes of anemia, clonal disorders of the hematopoietic stem and progenitor cells (HSPCs), including myelodysplastic syndromes or neoplasms (MDS), are possible differential diagnoses.
Over the last few years, the identification of clonal hematopoiesis (CH) has been facilitated by the advent of next-generation sequencing (NGS) in our clinical practice, which is challenging the sharp distinction between age-associated benign CH and overt neoplastic conditions.3 In most patients with unexplained cytopenia, a bone marrow examination is required to identify or exclude potential causes. The diagnosis of MDS has recently been revised by two competing classification systems.4–6 These are based on the previous World Health Organization (WHO) classification and include novel genetically defined entities such as MDS with mutations in SF3B1 (associated with ringed sideroblasts [RS]) and biallelic mutations in TP53. The transition to secondary acute myeloid leukemia (AML) is increasingly defined by genetic markers and not exclusively by blast counts ≥20%. In contrast to the increasing relevance of molecular diagnostics, the distinction of unclear cytopenia from MDS remains defined exclusively by the presence of morphological dysplasia. However, some patients with unclear cytopenia may not reach the required ≥10% dysplastic cells within a lineage or MDS-defining cytogenetic aberrations. Therefore, it remains a major drawback that assessment of dysplasia is subjective with high inter-observer variability, affecting the distinction of clonal cytopenia of undetermined significance (CCUS) and MDS.7,8 Cases with persistent cytopenia for more than four to six months and without any other clinical condition explained by bone marrow assessment can be classified as idiopathic cytopenia of undetermined significance (ICUS).9 However, if the presence of clonality has not been ruled out by sensitive methods, an underlying hematological condition cannot be faithfully excluded.
Nowadays, NGS is frequently used to complement cytogenetic analyses for the assessment of patients with cytopenia. Different technologies are currently used in the clinical setting to identify somatic leukemia-associated driver mutations (SLADMs) in genes involved in RNA-splicing, epigenetic regulation, DNA transcription, cell cycle regulation and cell signaling (Table 1).10 These technological advances have substantially facilitated the timely identification of clonality but the management of these patients remains challenging. Clonal hematopoiesis of indeterminate potential (CHIP) is defined by the presence of SLADMs at a variant allele frequency (VAF) ≥2% in individuals without cytopenia or other signs of hematological disease (Table 2). CHIP is a relevant phenomenon observed in the aging population, affecting 20–40% of individuals >80 years old, and associated with an increased risk for transformation to overt hematological malignancies, as well as other inflammatory-degenerative conditions, including cardiovascular disorders and chronic obstructive pulmonary disease (COPD).10–13
Patients with cytopenia not fulfilling the morphological or cytogenetic criteria of MDS but with SLADMs at a VAF ≥2% are classified as having CCUS.12 However, elderly patients with asymptomatic blood cytopenia are not systematically referred for further investigations and clonality as such may be missed. CH is frequent in elderly individuals and the borderline between random alterations of the elderly, non-neoplastic immune-selected conditions (aplastic anemia, paroxysmal nocturnal hemoglobinuria) and overt myeloid malignancies (MDS) are increasingly blurred. This review provides a structured approach to the assessment of unclear cytopenia and summarizes the clinical implications of CH, as well as our current management of CCUS patients.
Diagnostic approach in patients with unclear cytopenia
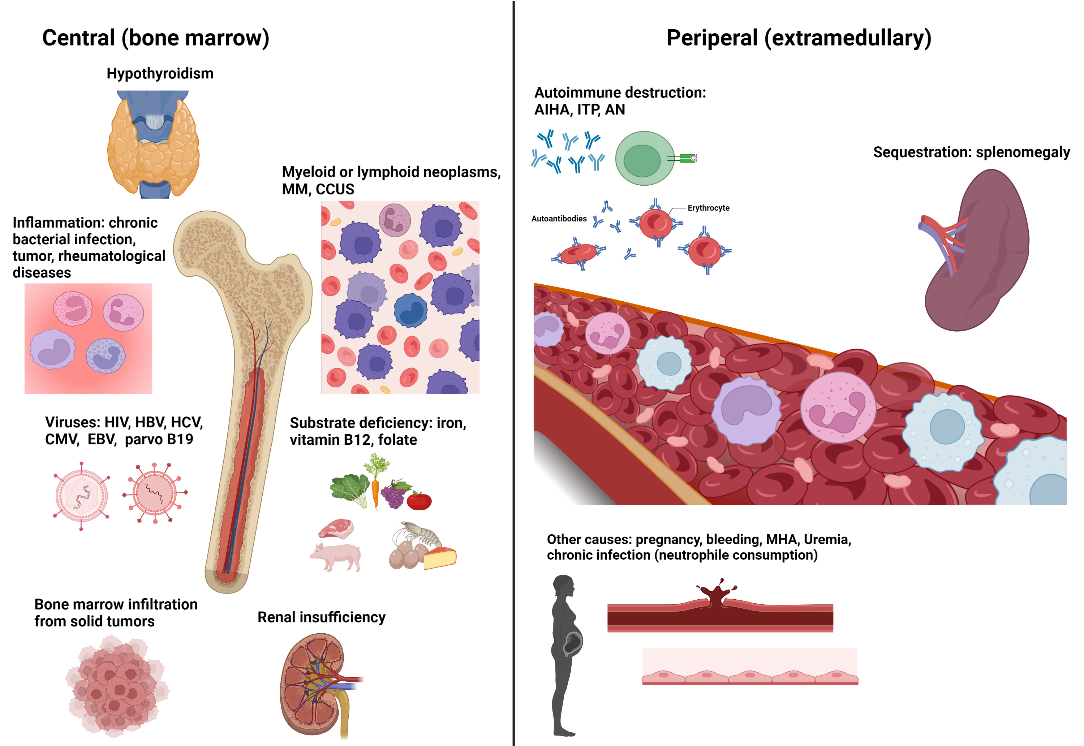
Potential causes of cytopenias and diagnostic work-up are summarized in Figure 1 and Figure 2, respectively.
.png)
.png)
Basic diagnostic work-up
A thorough medical history and physical examination for signs of bleeding, hepatosplenomegaly, lymphadenopathy and inflammatory conditions is the cornerstone of the initial clinical assessment. This may reveal hematological and non-hematological causes of anemia or other cytopenias. Possible causes comprise substrate deficiencies (iron, folate, vitamin B12), reactive hyporegeneration (renal insufficiency, infection, inflammation, hypothyroidism), peripheral consumption (bleeding, hemolysis, immune thrombocytopenia or neutropenia), splenic sequestration (liver diseases, splenomegaly), displacement of hematopoiesis by hematological and non-hematological disorders (myeloma, lymphoma and lymphoproliferative disorders, fibrosis, bone marrow infiltration from solid organ tumors), as well as intrinsic defects of the hematopoietic stem and progenitor cells (HPSCs) (myeloid neoplasia). To this end, laboratory work-up should include a complete automated blood cell count with the differentiation of white blood cells, reticulocytes, red blood cells and thrombocyte indices.
Cytomorphological examination of the peripheral blood smear can be instructive for identifying morphological abnormalities of red blood cells (like anisocytosis, poikilocytosis, schistocytes, tear-drops), atypical or dysplastic features of white blood cells or even immature precursors. Furthermore, the laboratory testing should include serum iron, transferrin, transferrin saturation, ferritin, serum folate, transcobalamin, vitamin B12, creatinine, C-reactive protein (CRP), thyroid-stimulating hormone (TSH), lactate dehydrogenase (LDH), bilirubin, haptoglobin, alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT). Direct antiglobulin test, antinuclear antibodies (ANA), antineutrophil cytoplasmic antibodies (ANCA), rheumatoid factor (RF), neutrophil and platelet antibodies can be useful if an underlying autoimmune process is suspected.
Serologies for the most common chronic viral infections associated with cytopenias are also useful, including human immunodeficiency virus (HIV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), hepatitis B virus (HBV), hepatitis C virus (HCV) and parvovirus B19. Serum protein electrophoresis with immunofixation and free light chain assays can be indicative of lymphoid or plasma cell neoplasms. An abdominal ultrasound should be part of the initial evaluation to identify liver cirrhosis, splenomegaly, intraabdominal lymphadenopathies or tumors that may not be detectable by clinical examination.
Advanced diagnostic work-up
If cytopenia cannot be fully explained by any extra-medullary condition, all patients that are sufficiently fit for further investigations and potential treatments should be referred to a hematologist. The selection of these patients remains a matter of debate and should be individualized based on the age, comorbidities, functionalities and wish of the patient. More specialized diagnostic work-up includes morphological evaluation of the peripheral blood, as well as bone-marrow with cyto- and histomorphological assessment, flow cytometry, cytogenetics and molecular analyses (https://mds-europe.org/management).14 Morphological examination of the peripheral blood smear can provide some indication of potential underlying disorders. Some examples are the presence of macrocytosis of the red blood cells in case of myelodysplastic or bone marrow failure syndromes, nuclear abnormalities and/or dysgranulopoiesis in the neutrophils or the platelets in MDS, blasts or myeloid precursors in myeloid neoplasms and the presence of atypical lymphocytes in lymphoproliferative disorders.15 Bone marrow cytomorphology and histopathology are essential for the identification of the cellularity of the bone marrow (bone marrow failure), disruptions in the quantity and quality of affected blood cell lineages (MDS), as well as displacement of hematopoiesis by hematological (leukemia, lymphoma, myeloma) and non-hematological conditions (fibrosis, metastatic cancer). The finding of dysplastic changes ≥10% in at least one hematopoietic cell lineage, ≥15% RSs or excess of blasts ≥5% may be sufficient for the morphological diagnosis of MDS.4 Hypocellularity and fibrosis play an additional important role in the new classification system.5,6 However, in some cases, morphological features are not sufficiently prominent or they can be misleading in reactive conditions (medications, underlying infectious or rheumatological diseases) for an affirmative diagnosis of MDS. In such situations, cytogenetic aberrations may help in detecting MDS-defining alterations and can lead to the diagnosis of MDS.4 This includes a complex karyotype (≥3 abnormalities), deletion or loss due to unbalanced translocation of 5q, 7q, 12p and 17p, monosomy 7, 11q deletion, monosomy 13 or 13q deletion, isochromosome 17q and isodicentric X chromosome (idic(X)).6
Cytogenetic alterations have implications for MDS risk stratification.16 However, some of the cytogenetic alterations may be signs of clonality but are not sufficient for the affirmative diagnosis of MDS. If the above-described examinations are not diagnostic, molecular analyses using NGS can be considered in most cases. We generally advice to seek for a confirmation of cost coverage by the health insurance before testing. Molecular markers are increasingly used in all patients undergoing bone marrow examinations as they provide additional clues for clonality, more refined prognostic value (International Prognostic Scoring System-Molecular [IPSS-M]) and can be used as measurable markers for disease follow-up.17,18 Especially in younger, transplant-eligible patients, molecular diagnostics should always be part of the initial specialized assessment.19 NGS not only improves prognostic information (i.e., bi-allelic TP53, EZH2, ETV6, RUNX1, ASXL1, SRSF2, U2AF1, RAS-pathway and JAK2 with VAF ≥2%) but is also instrumental to identify clonality in patients with unclear cytopenia, sub-classify patients by the occurrence of specific mutation patterns (i.e., SF3B1 in RS-MDS subtypes, SRSF2 in chronic myelomonocytic leukemia [CMML], JAK2 in myeloproliferative neoplasms [MPN], KIT in advanced systemic mastocytosis [ASM], FLT3/NPM1 in AML) and represent a baseline for a subsequent assessment of minimal residual disease (MRD) after allogeneic hematopoietic stem cell transplantation (allo-HSCT). For patients with unclear cytopenia, some characteristic molecular features (i.e., VAF >10%, spliceosome-mutations, DTA-plus, multiple mutations) behave as an overt hematological neoplasm.20 These molecular features can be used for the distinction of hypoplastic MDS from aplastic anemia.21 Incomprehensibly, these molecular patterns have not found their way into the current MDS classification, where the distinction between MDS and CCUS remains mainly based on morphology and cytogenetics with exception of SF3B1. These inconsistencies should be revised in the future classification systems.
Clinical implications of clonal cytopenia of undetermined significance
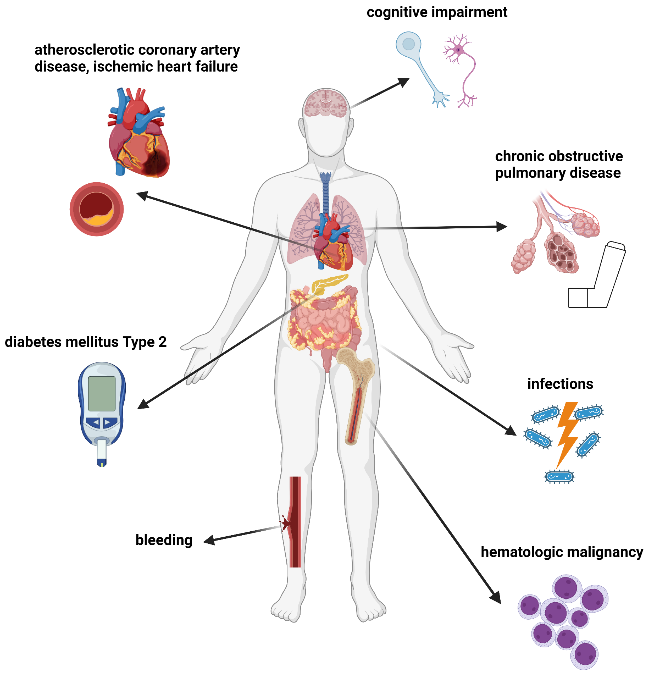
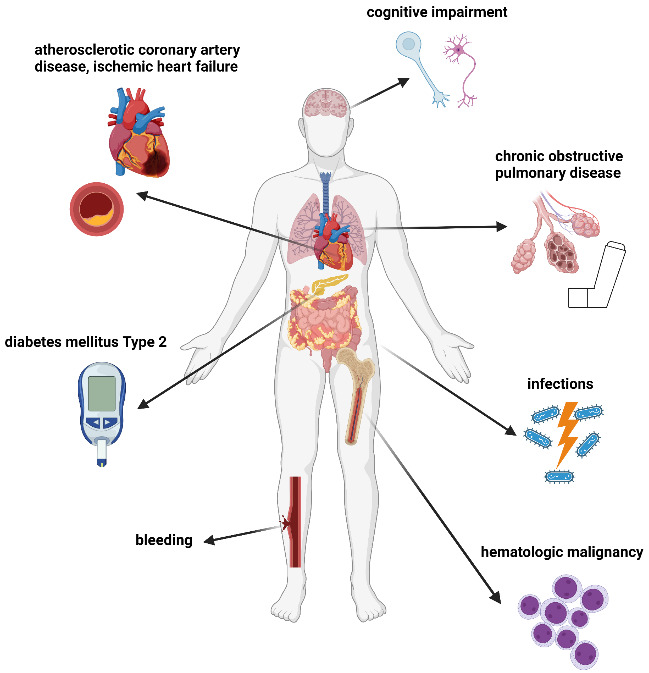
Clinical implications in CH and CCUS are summarized in Figure 3.22
Risk of subsequent myeloid neoplasms
ICUS can be considered a benign or (pre)malignant condition, which may be discernible by further molecular analyses.9 Depending on the absence or the presence of clonality in ICUS/CCUS, the 5- and 10-year cumulative probabilities of progression to myeloid neoplasms are 9% vs 82% and 9% vs 95%, respectively.20 Generally, higher VAFs (>10%), mutations in spliceosome genes (i.e., SF3B1, SRSF2, U2AF1) and more than one mutation represent a higher risk for an overt hematological neoplasm (Figure 4).

Cardiovascular diseases
Patients with peripheral blood cytopenia, whether clonal or not, are at increased risk of cardiovascular morbidity and mortality compared to age-controlled groups, which cannot be explained by their traditional cardiovascular risk factors.23 In mouse models, somatic mutations in the TET2, DNMT3A and JAK2 genes have been shown to contribute to atherosclerosis, as well as myocardial remodeling through increased inflammation promoted by clonally affected immune cells.24
Chronic obstructive pulmonary disease
In a recent study, a positive correlation between CHIP and COPD severity was found in four patient cohorts using whole genome and exome sequencing.25 In vivo, accelerated emphysema development was observed in mice with TET2 deletion in the hematopoietic compartment exposed to cigarette smoke.
Metabolic diseases
Defects in metabolic pathways associated with type II diabetes or hypercholesterolemia seem to induce the proliferation of HPSCs and lead to monocytosis, whereas in murine models, a TET2 mutation-driven CHIP exacerbated obesity and insulin resistance.26–28
Cognitive impairments
The Women’s Health Initiative Memory Study was a multicenter, randomized, double-blind, placebo-controlled trial that investigated the effects of hormone therapy use in postmenopausal women on dementia and mild cognitive impairment.29 A subgroup of women without a history of a cerebrovascular event and a baseline blood sample for whole genome sequencing underwent annual neurocognitive assessments for up to 22 years to evaluate possible associations between CHIP and cognitive outcomes (n=934). No correlation between CHIP and the risk of developing a cognitive impairment was observed in a whole genome sequencing analysis. However, a risk stratification analysis by mutations in CHIP driver genes showed an elevated but not statistically significant risk for women with TET2 mutation.30
Systemic inflammatory and autoimmune manifestations (SIAMS)
Chronic inflammatory and autoimmune manifestations are frequently observed in patients with clonal hematopoiesis, as well as myeloid neoplasms, and patients with chronic autoimmune disorders are at an increased risk of developing hematological malignancies.31,32 It is currently unknown whether autoimmunity is a manifestation or a disease-driving factor towards myeloid malignancies. Increased levels of cytokines (interleukin [IL]-1β, tumor necrosis factor [TNF]-α, IL-6) and important effectors of the innate immune system (Toll-like receptors [TLRs], inflammasome) play a pivotal role in the development and progression of myeloid neoplasms.33–35 Their activation leads to a hyperinflammatory state leading to specific cell death, called pyroptosis, of HSPCs.35,36 Similar observations are made in several ICUS conditions like refractory anemia, chronic idiopathic neutropenia and chronic amegakaryocytic thrombocytopenia.37 Studies have found an association of CHIP in patients with autoimmune or inflammatory-degenerative conditions, such as systemic sclerosis and ulcerative colitis, or in patients undergoing total hip arthroplasty.38–40
Clonal dominance
Most individuals harbor CH if high-sensitivity assays with error-corrected sequencing technologies are applied.41 Increasing evidence from basic research supports the notion that cell-intrinsic combined with cell-extrinsic factors, such as inflammation, shapes the selection of clonal at the expense of normal HSCPs.42 This is based on the observation that normal HSPCs are exhausted by excessive differentiation in an inflammatory microenvironment, whereas clonal HSPCs seem to keep a myeloid-biased stem cell function.43 Moreover, CHIP clones have shown a higher repopulation capacity in the context of autologous HSCT44 and the use of CHIP donors seems safe in allo-HSCT.45 Moreover, different mutations may have gene-specific fitness effects in clonal dominance,46 however, the differential impact of CHIP mutations and their co-occurrence needs further investigation.
Management of patients with clonal hematopoiesis
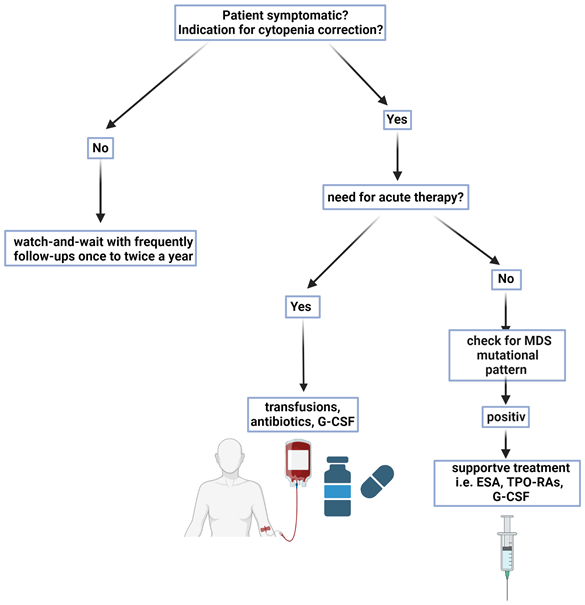
Management of patients with CH and CCUS is summarized in Figure 5. Cytopenia of unknown origin with/or without clonality that does not fulfill the formal criteria for bone marrow failure or an overt myeloid malignancy represents a challenge regarding further management and international consensus guidelines are currently under development. There are no prospective studies that determine the efficacy or safety of early clinical interventions for associated conditions and targeted therapies to prevent clonal evolution.

Counseling with “watch and wait”
“Watch and wait” is the most frequent approach in asymptomatic patients. However, as the risk of patients with CCUS for developing myeloid neoplasia, cardiovascular complications and other inflammatory-degenerative conditions is increased, a thorough counseling and follow-up management plan should be in place. The potential risks need to be explained and overuse of the health system resources has to be avoided. The individual quantification of risk is difficult, as comorbidities and comedications may represent relevant cofactors and influence the thresholds for further investigations (anemia in patients with cardiovascular or pulmonary disorders, thrombocytopenia in patients with anticoagulation or platelet anti-aggregation). The specialized diagnostic work-up should be repeated as soon as clinical or laboratory signs of deterioration occur.
Reduction of cardiovascular risk factors
Screening for modifiable cardiovascular risk factors should be offered for smoking, overweight, diabetes, hypertension and hyperlipidemia. The quantification of the long-term risk with established score calculators currently does not account for the additional risk associated with CH and the risks may differ depending on comorbidities and treatments.47 Close communication with the primary care physician remains a cornerstone to establishing a management plan aiming for lifestyle modification to increase exercise, reduce overweight and stop smoking. Some patients may require drugs to control hypertension, dyslipidemia and diabetes mellitus according to published guidelines. Unfortunately, non-adherence to guidelines regarding cardiovascular risk reduction is frequent in the primary care setting and should be taken into account.48,49 Therefore, an active investigation of the above-mentioned goals should be an integral part also for hematological follow-up visits.
Supportive treatment
Anemia, especially in the elderly, is associated not only with reduced physical function but also with cognitive dysfunction and increased morbidity and mortality.2 The incidence of patients with severe anemia, thrombocytopenia or neutropenia is not well established in CCUS and should call for careful assessment for the presence of overt MDS or another myeloid neoplasm, which will allow access licensed treatments.
Therapy with erythropoiesis-stimulating agents (ESA) is well established in patients with low-risk MDS and in those with anemia associated with chronic kidney disease, leading to a better quality of life (QoL), as well as delay/reduction of transfusions. In contrast, data in the context of CCUS are scant. Erythropoietin (EPO) levels seem to be inappropriately low in the anemia of unknown origin (AUO) in the elderly, suggesting insufficient EPO production in response to anemia.50 A study comparing epoetin alfa versus placebo in a cohort of mainly African-American women with AUO aged ≥65 years showed a significant increase in hemoglobin (Hb) levels and patients’ QoL with epoetin alfa.51 However, there is an unmet need for larger studies to examine the effect of ESA therapy in AUO, ICUS and CCUS patients.
Patients with mild to moderate thrombocytopenia with no relevant bleeding (thrombocytes >50,000/μL) usually do not need therapy and observations with regular follow-up visits may be sufficient. The risk of bleeding usually increases when thrombocyte counts fall below 10,000/μL, but occasional bleeding can also occur at higher values due to a concomitant dysfunction. A history of prior bleeding, thrombocyte counts <20,000/μL and an age of >60 years increase the bleeding risk. Prophylactic platelet transfusions should be considered at thrombocyte count <10−20,000/μL, depending on the individual risk of bleeding.52,53 In these cases, an initial steroid trial, analog to the treatment recommendations for immune thrombocytopenia (ITP), may be tried after a full diagnostic assessment.54 No data on efficacy and safety is available on the use of thrombopoietin receptor agonists (TPO-RAs) in ICUS and CCUS, however, some of those patients may be currently misclassified as ITP patients, if a molecular assessment was not done. However, both conditions may co-occur and it remains difficult to estimate the individual relevance of each component to the occurrence of thrombocytopenia.19
Regarding the management of neutropenia, the individual risk of infection should be considered. A diminished production in the bone marrow, an absolute neutrophil count (ANC) of <500/μL and a history of recurrent or severe infections indicate an increased risk. In these cases, preventive measures like hand and dental hygiene, and regular review of the vaccination plan (influenza, COVID-19, pneumococci and varicella zoster virus [VZV]) may be warranted for most of these patients that are ≥65 years old. Evaluation of a pre-emptive treatment with granulocyte colony-stimulating factor (G-CSF) remains controversial, even in MDS. Multicenter trials investigating the prophylactic use of G-CSF versus no-treatment in neutropenic patients showed a significant reduction in the incidence and duration of severe bacterial infections as well as a reduced duration of antibiotic therapy, however, specific studies in this context are lacking for MDS and CCUS patients.55,56 In case of clinical suspicion of an acute infection in patients with ANC <800/μL and inadequate neutrophil production in the bone marrow should receive immediate broad-spectrum antibiotics with coverage of Gram-negative bacteria after obtaining blood and urine samples for cultures. A great proportion of these patients do not exhibit the typical signs of infection and fever of unknown origin can be the only clinical sign. Prophylactic treatment with antibiotic or antifungal therapy in non-chemotherapy-induced neutropenia has not demonstrated any relevant effect on the incidence, severity and duration of infections and this may also hold for MDS patients.57
Approach to systemic inflammatory and autoimmune manifestations
Patients with myeloid neoplasms have a higher incidence of SIAMs. The contribution of patients with CH remains to be established but from our clinical experience, we have identified an increasing number of patients with unclassifiable SIAMs that harbor CH, CCUS or overt MDS. Therefore, a close collaboration with immunologists and rheumatologists is advisable for those patients as well as the search for the pathognomonic somatic mutation in the UBA1 gene for VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic).58,59 Polychondritis, Sweet syndrome and neutrophilic dermatoses have a high prevalence in VEXAS, which has an association with myeloid or lympo-/plasmocytic neoplasms.60 Autoinflammatory manifestations in this novel autoinflammatory disorder can be very diverse and may respond to high doses of steroids but relapse at tapering.61 Responses to steroid-sparing drugs have been reported with IL-1, IL-6, TNF-α and JAK-STAT signaling inhibition, however, some of those patients remain refractory and are candidates for allo-HSCT.62 In our VEXAS cohort, we have identified surprisingly high VAFs in TET2 and DNMT3A, which may indicate a selective advantage of clonally affected HSPCs in the inflammatory context.63
Moreover, many patients suffer also from unclassifiable inflammatory cardiovascular (excessive atherosclerosis and degeneration of grafts), dermatological (neutrophilic dermatoses) and pneumological (excessive inflammatory destructive COPD and interstitial lung disease) manifestations. We have just started to scratch the surface of understanding the biological interplay between clonally-affected immune cells with age-related degenerative-inflammatory conditions. Due to the complexity and heterogeneity of manifestations, an interdisciplinary care team embedded in a translational research environment is of utmost importance for the management of those patients.
Follow-up
Taking the risk of subsequent myeloid neoplasms into account, especially in CCUS patients with high-risk predictive mutations, a careful follow-up plan should be discussed with the patient. The patients’ expectations, life expectancy and comorbidities need to be considered. In younger patients, every deterioration of blood counts and constitutional or inflammatory symptoms should prompt a new diagnostic work-up with peripheral blood and bone marrow assessment. In patients with constitutional and inflammatory symptoms, also a PET scan with targeted biopsies may be of value to exclude potential other reasons for constitutional symptoms (lymphoma, Castleman-like disease, hemophagocytosis) as well as an extra-medullary transformation.
Conclusions
With the aging of the general population, the incidence of CH and CCUS is expected to increase substantially. Given the associated risk of developing overt hematological malignancies and the potential negative impact on inflammatory-degenerative conditions of the elderly (cardiovascular disease, COPD, dementia, non-classified inflammatory conditions), CCUS deserves more attention in the future management of elderly individuals. As long as specific treatments are not available that prevent or abrogate clonal evolution and associated inflammatory conditions, establishing an adequate plan for further diagnostic evaluation, follow-up and prevention remain the only management options currently at hand. However, as age-associated disorders will continue to drive health care expenses, a better understanding, prevention, and potential early intervention remain unmet needs. Screening for CH should be recommended in all patients, in whom cytopenia is not sufficiently explained by non-hematological disorders or with unusual hyper-inflammatory conditions, especially for patients at a younger age or elderly patients that are not frail. Clinicians should identify those patients that are fit enough for further evaluation and potential treatment and should not refrain to refer those patients to hematologists for more specialized diagnostics. CH is a biological phenomenon that received growing clinical interests based on the advances in sequencing technologies, and, which is filling the gap between benign blood conditions in the elderly population and overt myeloid malignancies. As for now, it is relevant to identify those patients with relevant clonal patterns that harbor a higher risk of transformation to overt myeloid malignancies and to find potential explanations for unusual degenerative-inflammatory conditions that are associated with CH. As long as we do not have specific treatments licensed for CH, it will remain, from the perspective of reimbursement of health care services, unattractive to run specialized CHIP/CCUS clinics. For the moment, we are lacking prospective, controlled interventional data and are mainly relying on association studies and risk assessments. This might be relevant for biological understanding and patient counseling but not sufficient to base any intervention on robust data. As current research has made advances in identifying potential therapeutic targets, clinical trials in CH and CCUS are urgently needed to prevent clonal evolution towards myeloid neoplasm and abrogate hyper-inflammatory conditions. If these trials come up with effective treatments, with low toxicity, simple application schedules and reasonable costs, CCUS may become suitable for early interventions.
Conflict of interest
The authors declare no conflict of interest for this review. Potentially perceived conflicts of interests according to the definitions and terms of International Committee of Medical Journal Editors are JC: Gilead: financial support for travel; NB: Alexion: research funding to institution; Amgen: financial support for travel; Astellas: research funding to institution; Celgene/BMS: financial support for travel, research funding to institution; consultancy honoraria; Gilead: financial support for travel; Keros: consultancy honoraria; Janssen: financial support for travel; Novartis: financial support for travel, research funding to institution, consultancy honoraria; Roche: financial support for travel, research funding to institution; Sandoz: research funding to institution; Servier: research funding to institution; Takeda: research funding to institution.
Funding
Not applicable.
Authors contributions
Ioannis Chanias and Nicolas Bonadies wrote the review.