
Introduction
Antibody-drug conjugates (ADCs) have emerged as an effective class of antitumor therapeutics that exploit antibody specificity to deliver highly potent cytotoxic agents to cancer cells.1 Simply put, ADC technology involves conjugating an engineered monoclonal antibody (mAb) to a cytotoxic drug (known as the payload) via a versatile chemical linker. The classical ADC mechanism involves the antibody component specifically binding to the target antigen receptor that is highly expressed on the surface of tumor cells, thus minimizing systemic exposure and reducing side effects. Once internalized, the ADC-antigen complex relies on intracellular trafficking and processing by the endo-lysosomal system to release sufficient cytotoxic payload into the cell cytoplasm to trigger apoptosis.1,2
The design of a successful ADC is not without its challenges. For example, stability of the labile linker during storage and in the blood circulation is required to prevent the early release of free drug and the potential for off-target toxicity and other side effects.3 In most ADCs, the payload is designed to be released using either a cleavable or a non-cleavable linker.1 Conjugating a cytotoxic drug to random reactive groups on an antibody can produce heterogeneous ADC preparations with a range of drug antibody ratios (DAR), different physical stabilities and safety profiles.4 Extreme hydrophobicity of a cytotoxic agent can also change the biological properties of an antibody and lead to aggregation during conjugation or storage.3 Another major challenge involves obtaining the optimal concentration of the ADC preparation to enhance local bioavailability and tissue penetration.5 Second-generation ADCs were therefore designed with novel linkers to optimize biodistribution and payload delivery.
The effectiveness of ADCs in solid tumors has been limited in the past due to the heterogeneous expression of tumor-associated antigen receptors. Evidence from the literature indicates that heterogeneity in target antigen receptors and the distribution of ADCs in the tumor microenvironment can promote the selection of resistant tumor cells.6 However, preclinical studies have suggested that second-generation ADCs with cleavable peptide-based linkers may potentially kill adjacent antigen-negative tumor cells, a process referred to as bystander killing.7 ADCs with bystander effects, where the payload can diffuse out of the target cell into the adjacent tumor microenvironment, have the potential to improve clinical efficacy and treatment outcomes for patients with solid tumors. Indeed, significantly improved survival outcomes were shown with trastuzumab deruxtecan, a human epidermal growth factor receptor 2 (HER2)-directed ADC, in patients with HER2-low metastatic breast cancer, irrespective of hormone receptor (HR) status, in the pivotal phase III DESTINY-Breast04 trial.8 Positive results were also reported with sacituzumab govitecan, a trophoblast cell surface antigen 2 (Trop-2)-directed ADC, in patients with metastatic triple-negative breast cancer (mTNBC), regardless of tumor Trop-2 expression level.9 Although greater efficacy was observed in those with a medium or high Trop-2 score, also patients with low Trop-2 score achieved improved outcomes when treated with sacituzumab govitecan versus chemotherapy, but the small number of patients in this subgroups limited definitive conclusions. Due to the current lack of extensive evidence, understanding the interplay between antigen heterogeneity, bystander activity and heterogeneous delivery of the payload to adjacent cells remains to be further elucidated.
With recent advances in bioengineering technology, this class of drugs is gaining momentum for the targeted treatment of solid tumors, as well as other cancer types. This review article provides an overview of the current clinical landscape of ADCs targeting solid tumors, including breast, colorectal, lung, prostate, ovarian and urothelial cancers.
Current clinical landscape of ADCs in solid tumors
Several ADCs have received approval to treat various solid tumors from the Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Table 1 provides a summary of these commercially available ADCs. In addition to FDA and EMA approval, three ADCs, trastuzumab emtansine,10 trastuzumab deruxtecan11 and sacituzumab govitecan,12 are also approved in Switzerland to treat adults with HER2-positive breast cancer and triple-negative breast cancer (TNBC), respectively. In addition, enfortumab vedotin was recently approved in Switzerland for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who have received platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting and who have experienced disease progression or relapse during or after treatment with a programmed cell death receptor-1 (PD-1) or programmed death ligand-1 (PD-L1) inhibitor.13
Trastuzumab emtansine
Trastuzumab emtansine was the first ADC approved in solid tumors, based on findings from the EMILIA study.22 EMILIA was a phase III randomized study evaluating trastuzumab emtansine versus standard therapy with capecitabine and lapatinib. The trial included 991 HER2-positive metastatic or locally advanced breast cancer patients who had previously received trastuzumab and a taxane. At a median follow-up of 19 months, results showed a clear efficacy benefit for trastuzumab emtansine over standard therapy (Table 2). In the final analysis, the incidence of grade ≥3 adverse events (AEs) was 48% in the trastuzumab emtansine group and 60% in the control group.14 The most frequent grade ≥3 AEs with trastuzumab emtansine were thrombocytopenia (14%), increased transaminases (8%) and anemia (4%). Among patients treated with capecitabine and lapatinib, diarrhea (21%), palmar-plantar erythrodysesthesia syndrome (18%) and vomiting (5%) were the most common grade ≥3 AEs.
At that time, the EMILIA trial efficacy results were unprecedented in metastatic breast cancer. In 2019, trastuzumab emtansine was also indicated in early breast cancer for HER2-positive women who have residual disease in the breast and/or lymph nodes after preoperative taxane-containing chemotherapy in combination with at least trastuzumab as HER2-targeted therapy.10 This was based on the results from the phase III, open-label KATHERINE trial, which demonstrated significantly improved invasive disease-free survival among patients receiving trastuzumab emtansine versus trastuzumab (HR: 0.50 [95% CI: 0.39−0.64]; p<0.001), after a median follow-up of approximately 41 months.15 At 3 years, the estimated percentage of patients who were free of invasive disease was 88.3% with trastuzumab emtansine and 77.0% with trastuzumab. The safety data were consistent with the known safety profile of trastuzumab emtansine (T-DM1).
Sacituzumab govitecan
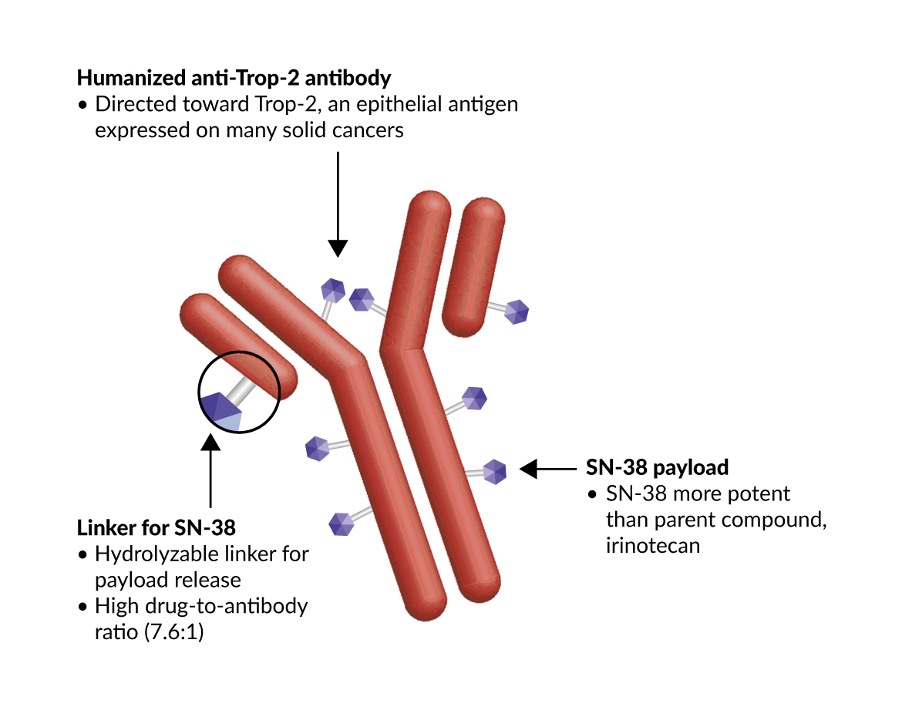
The second ADC to gain regulatory approval in Switzerland is the first-in-class Trop-2-directed antibody and topoisomerase inhibitor conjugate, sacituzumab govitecan. In the landmark ASCENT study, sacituzumab govitecan was the first ADC that showed improved clinical outcomes in patients with relapsed or refractory mTNBC who had received at least two prior lines of systemic therapies.18 TNBC is a highly heterogenic and aggressive breast cancer subtype characterized by tumor cells that lack HER2, estrogen and progesterone receptors.23 It accounts for about 15–20% of diagnosed breast cancers and typically affects younger women.23,24 With limited treatment options, the prognosis is poor for patients with advanced or metastatic disease, with a median survival of approximately 12 months.25 Unlike trastuzumab emtansine and other ADCs, sacituzumab govitecan has a novel hydrolyzable CL2A linker that may contribute to a bystander effect on neighboring cancer cells regardless of HER2 expression levels (Figure 1).26 The linker is particularly susceptible to pH changes, which may permit the release of the SN-38 payload both internally in an acidic environment of the tumor cell and at a lower pH in the tumor microenvironment. The high ratio of drug to antibody (DAR, 7.6) enables delivery of high concentrations of the SN-38 payload to tumors.27
-2-directed_antibody-dru.jpg)
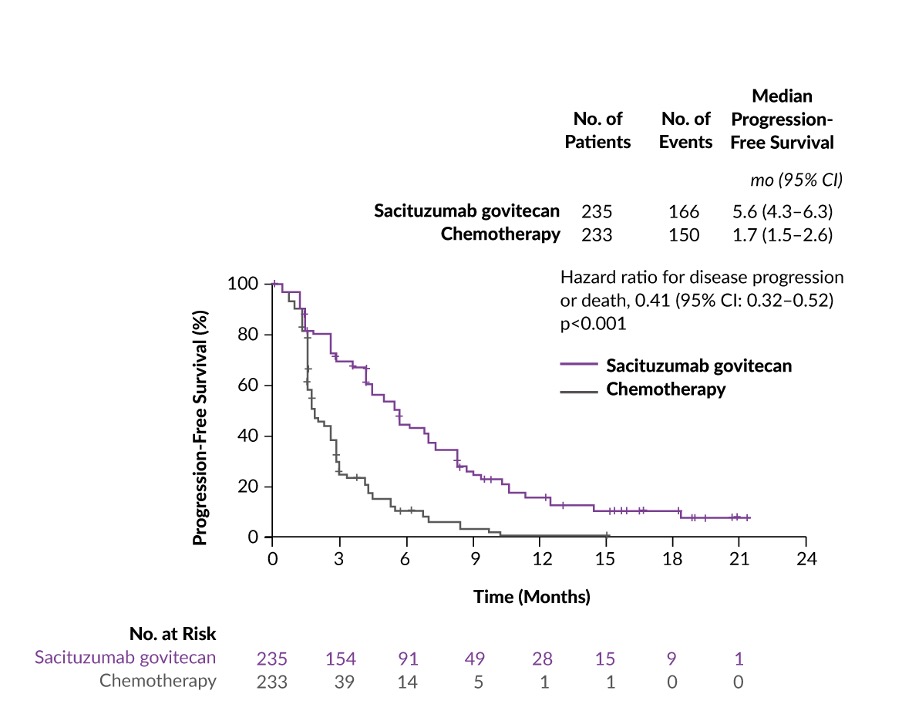
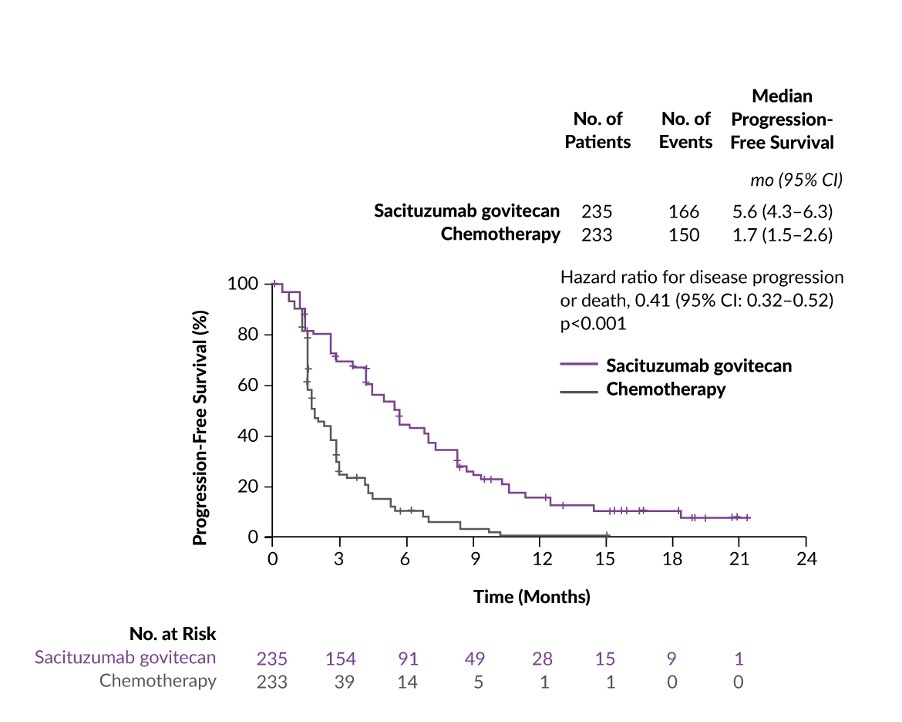
ASCENT was a phase III confirmatory, multicenter, randomized controlled trial of sacituzumab govitecan versus single-agent chemotherapy treatment of physician’s choice (TPC) conducted in 529 chemotherapy-pretreated mTNBC patients.18 At baseline stratification, 61 patients had brain metastases and 468 patients had no history of brain metastases. In patients without brain metastases, the median progression-free survival (PFS) (primary endpoint of the study) was 5.6 months for those treated with sacituzumab govitecan (n=235) and 1.7 months for those treated with TPC (n=233) (HR: 0.41; p<0.001) (Figure 2). Similarly, the median OS in this patient population was nearly doubled with sacituzumab govitecan therapy compared with TPC chemotherapy (12.1 months vs 6.7 months; HR: 0.48; p<0.0001). Furthermore, the overall response rate (ORR) in the sacituzumab govitecan arm was 35% compared with 5% in the chemotherapy arm. In terms of safety, the most common grade ≥3 treatment-related AEs were neutropenia (51% with sacituzumab govitecan vs 33% with chemotherapy), leukopenia (10% vs 5%), diarrhea (10% vs <1%), anemia (8% vs 5%) and febrile neutropenia (6% vs 2%). Among patients receiving sacituzumab govitecan, the incidence of rash and ocular toxic effects was low and no neuropathy of grade >2 was reported. Notably, no grade 1 or 2 interstitial lung disease (ILD) occurred; grade 3 pneumonitis developed in 1 patient.
More recent data from the ASCENT study further showed that in relapsed or refractory mTNBC patients with at least two prior lines of therapy, sacituzumab govitecan maintained or improved health-related quality of life (HRQoL).29 For the primary HRQoL domains of global health status/QoL, physical functioning, role functioning, fatigue and pain, the sacituzumab govitecan arm showed statistically significant and/or clinically meaningful greater improvements than the TPC arm. Sacituzumab govitecan also significantly prolonged time to first deterioration in all HRQoL domains except for global health status/QoL and significantly shortened time to improvement in physical functioning and pain.
In addition to its approval in mTNBC, sacituzumab govitecan has been recently approved in the US to treat locally advanced or metastatic urothelial cancer (mUC).30 This approval was based on data from the multicenter, open-label, phase II TROPHY-U-01 study, which assessed the efficacy and safety of sacituzumab govitecan in 113 patients with locally advanced or mUC after the failure of treatment with platinum-containing chemotherapy and either a PD-1 or a PD-L1 inhibitor.19 At a median follow-up of 9.1 months, sacituzumab govitecan demonstrated an ORR of 27%, with 77% of patients experiencing tumor shrinkage. The median duration of response (DoR) was 7.2 months, with median PFS and OS of 5.4 months and 10.9 months, respectively. Comparable with the safety data reported in the ASCENT study, the most common grade ≥3 treatment-related AEs with sacituzumab govitecan were neutropenia (35%), leukopenia (18%), anemia (14%), diarrhea (10%) and febrile neutropenia (10%).
Trastuzumab deruxtecan
Trastuzumab deruxtecan, an antibody-conjugate linked to a topoisomerase 1 inhibitor, was approved for the treatment of HER2-positive breast cancer based on the DESTINY-Breast01 trial.16 DESTINY-Breast01 is a phase II, open-label, multicenter, two-part trial that demonstrated impressive efficacy and durable responses of trastuzumab deruxtecan in patients with HER2-positive unresectable and/or metastatic breast cancer previously treated with trastuzumab emtansine (n=184). Saura Manich et al. (2021) recently presented the updated results from the DESTINY-Breast01 trial with an additional six months of follow-up data.31 At a median follow-up of 26.5 months, patients receiving trastuzumab deruxtecan continued to show an increase in response, with an updated ORR of 62.0% and a complete response (CR) rate of 7.1%. Median DoR was 18.2 months and the median PFS was 19.4 months. Regarding safety, most patients experienced an AE of any grade and 57.1% experienced a grade ≥3 treatment-emergent AE, most frequently decreased neutrophil count (20.7%), anemia (8.7%), nausea (7.6%), a decreased white cell count (6.5%), a decreased lymphocyte count (6.5%) and fatigue (6.0%).16 Overall, 13.6% of patients had ILD (grade 1–2: 10.9%; grade 3–4: 0.5%; grade 5: 2.2%).
The international phase III DESTINY-Breast03 trial further compared head-to-head trastuzumab deruxtecan with trastuzumab emtansine in the same patient population.32 In total, 524 patients with HER2-positive metastatic breast cancer, previously treated with trastuzumab and taxane, were randomized to receive either trastuzumab deruxtecan or trastuzumab emtansine. Trastuzumab deruxtecan demonstrated a highly statistically significant and clinically meaningful improvement in PFS, the primary endpoint, versus trastuzumab emtansine, with median PFS not reached versus 6.8 months, respectively (HR: 0.28; p=7.8x10-22). Differences in estimated 12-month overall survival (OS) rates were also observed at an early time point (94.1% with trastuzumab deruxtecan vs 85.9% with trastuzumab emtansine; HR: 0.56 [95% CI: 0.36–0.86]). After a median treatment duration of 14.3 months and 6.8 months, respectively, grade ≥3 treatment-emerged AEs occurred in 45.5% of patients treated with trastuzumab deruxtecan and 39.8% of patients treated with trastuzumab emtansine. The most common grade ≥3 treatment-related AEs in the trastuzumab deruxtecan group included neutropenia (19.1%), thrombocytopenia (7.0%) and nausea (6.6%). ILD/pneumonitis (8.2%) was the most frequent treatment-emergent AE associated with discontinuation of trastuzumab deruxtecan.
Enfortumab vedotin
mUC is notoriously difficult to treat since 70–80% of patients remain unresponsive to immune checkpoint inhibitor (ICI) therapies.33 Five-year survival in the metastatic setting for patients with bladder cancer is approximately 5%.34 Enfortumab vedotin is a first-in-class agent composed of an anti-nectin-4 monoclonal antibody attached to monomethyl auristatin E (MMAE), a microtubule-disrupting agent.35 The target of this ADC, nectin-4, is a cell adhesion molecule that is overexpressed on the surface of multiple cancers, including urothelial cancer (UC). In Switzerland, enfortumab vedotin was recently approved for patients with locally advanced or mUC who have previously received a PD-1/PD-L1 inhibitor and platinum-containing chemotherapy before or after surgery.13 The pivotal EV-301 study is a multicenter, phase III, open-label, randomized trial to evaluate enfortumab vedotin compared with physician’s choice of chemotherapy in 608 patients with locally advanced or mUC who were previously treated with a PD-1 or PD-L1 inhibitor and platinum-based therapies.36 The trial met its primary endpoint, with enfortumab vedotin significantly improving OS, resulting in a 30% reduction in risk of death (HR: 0.70 [95% CI: 0.56–0.89]; p=0.001). Enfortumab vedotin also significantly improved PFS, a secondary endpoint, with a 38% reduction in risk of disease progression or death compared with chemotherapy (median, 5.55 months vs 3.71 months; HR: 0.62 [95% CI: 0.51–0.75]; p<0.001). Rosenberg JE et al. (2021) recently published an analysis of hard-to-treat subgroups from the EV-301 trial, including age 65 years or older, presence of liver metastasis, primary upper tract disease and nonresponse to prior PD-1/PD-L1 inhibitor treatment.37 At a median follow-up of 11.1 months, OS was maintained across the majority of subgroups. For example, OS was 9.63 months versus 5.95 months (HR: 0.66 [95% CI: 0.456–0.957]) among patients with liver metastasis in the enfortumab vedotin and chemotherapy arms, respectively. Similar benefits were seen in terms of PFS and ORR in patients receiving enfortumab vedotin versus those receiving standard chemotherapy. Regarding safety, treatment-related AEs occurred in most patients (93.9% with enfortumab vedotin and 91.8% with chemotherapy), with a similar frequency of AEs of grade ≥3 between the two treatment groups (51.4% and 49.8%).36 The most common grade ≥3 AEs were maculopapular rash (7.4%), fatigue (6.4%) and decreased neutrophil count (6.1%) in the enfortumab vedotin group and decreased neutrophil count (13.4%), anemia (7.6%) and decreased white cell count (6.9%) in the chemotherapy group.
Tisotumab vedotin
For patients with recurrent or metastatic cervical cancer (mCC), treatment options are limited, with low response rates (13–50%) and a median OS of 6.5–16.8 months.38 Tisotumab vedotin is an ADC that includes an antibody targeting tissue factor (TF), a protein involved in tumor signaling and angiogenesis, conjugated with MMAE. The efficacy of tisotumab vedotin was evaluated in the innovaTV 204 trial, an open-label, multicenter, single-arm phase II trial.21 The trial included 101 women with recurrent or metastatic squamous cell, adenocarcinoma or adenosquamous cervical cancer whose disease had progressed with or after doublet chemotherapy with bevacizumab (if eligible by local standards) and who had received two or fewer previous systemic regimens for recurrent or metastatic disease. The confirmed ORR was 24% and included 7 CRs and 17 partial responses (PRs). The disease control rate was 72% and the DoR was 8.3 months. The median PFS was 4.2 months, with a 6-month PFS rate of 30%. The median OS was 12.1 months. Grade ≥3 treatment-related AEs were reported in 28% of patients treated with tisotumab vedotin and included neutropenia (3%), fatigue (2%), ulcerative keratitis (2%) and peripheral neuropathies (2% each with sensory, motor, sensorimotor and neuropathy peripheral). Serious treatment-related AEs occurred in 13% of patients, most commonly peripheral sensorimotor neuropathy (2%) and pyrexia (2%). One patient died due to septic shock considered to be related to therapy. Results from this study suggest that tisotumab vedotin may be a new therapy for pretreated patients with mCC and a confirmatory phase III trial comparing tisotumab vedotin and physician’s choice chemotherapy is ongoing.39
More recently, interim results from two cohorts of the phase Ib/II, multi-cohort, open-label innovaTV 205 trial of tisotumab vedotin in recurrent or mCC became available.40 Promising, durable antitumor activity was observed with tisotumab vedotin in combination with carboplatin as first-line therapy for patients with advanced cervical cancer who had not received prior systemic therapy, and in combination with pembrolizumab for patients with advanced cervical cancer who experienced disease progression after 1–2 lines of prior systemic therapy. At a median follow-up of 4.8 months, the ORR was 55% in the tisotumab vedotin/carboplatin cohort (n=33), including 2 CRs (6%) and 16 PRs (48%). The incidence of grade ≥3 AEs was 78.8%, including 57.6% related to tisotumab vedotin. Serious AEs occurred in 42.4% of patients; 15.2% were associated with tisotumab vedotin. Ocular events, bleeding and peripheral neuropathy were the most frequent AEs of special interest and were mainly of grade 1–2. At a median follow-up of 10.2 months, the ORR was 35% in the tisotumab vedotin/pembrolizumab cohort (n=34), including 2 CRs (6%) and 10 PRs (29%). In this cohort, grade ≥3 AEs occurred in 74.3% of patients; of these, 45.7% experienced AEs related to tisotumab vedotin. The rate of serious AEs was 42.4%, with 15.2% being tisotumab vedotin-related. Again, ocular events, bleeding and peripheral neuropathy were among AEs of special interest and were mainly of grade 1–2. Tisotumab vedotin is currently approved in the US for adult patients with recurrent or mCC with disease progression on or after chemotherapy.41
Ongoing trials with ADCs in solid tumors
Significant advances in bioengineering, bringing together a combination of potent payloads, better linker technology and better-identified tumor targets, are transforming the ADC space and clinical landscape for solid tumor patients.1 With approximately 82 novel ADCs in 150 active clinical trials registered with ClinicalTrials.gov for cancer patients, a plethora of ADCs are showing promise in phase I and II trials to meet unmet needs in a wide range of solid tumor types. Table 3 lists some such examples of ongoing trials with next-generation ADCs in various solid tumors, and emerging data appears promising.21,42–56
Trastuzumab duocarmazine is composed of a recombinant HER2 monoclonal antibody linked via a cleavable linker to the duocarmycin prodrug with potential antineoplastic activity.42 This novel ADC was investigated in heavily pretreated HER2-positive metastatic breast cancer patients in the two-part phase I clinical trial, SYD985.43 The primary outcome for the phase III SYD985.002/TULIP trial were presented at the European Society for Medical Oncology (ESMO) Congress 2021.42 The TULIP trial is an international, randomized superiority study comparing trastuzumab duocarmazine with TPC in 437 patients with pretreated HER2-positive locally advanced or metastatic breast cancer treated with two or more previous therapies or previous treatment with trastuzumab emtansine. The median number of prior lines of therapy was 4.0. Treatment with trastuzumab duocarmazine significantly improved PFS, the primary endpoint, compared with standard TPC, with a centrally-reviewed median PFS of 7.0 months and 4.9 months for trastuzumab duocarmazine and TPC, respectively (HR: 0.64 [95% CI: 0.49–0.84]; p=0.002). No significant differences were observed in ORR or HRQoL between the two treatment groups. In terms of safety, most patients experienced at least one treatment-emergent AE, with comparable rates of grade ≥3 events between the two treatment groups (52.8% with trastuzumab duocarmazine and 48.2% with TPC). The most common grade ≥3 treatment-emergent AEs with trastuzumab duocarmazine were keratitis (12.2%), conjunctivitis (5.6%) and neutropenia (4.9%). Among patients receiving TPC, neutropenia occurred in 18.2%, palmar-plantar erythrodysesthesia syndrome in 3.6% and diarrhea in 1.5%. Eye toxicity was reported in 78.1% of patients receiving trastuzumab duocarmazine (grade ≥3: 21.2%). In total, 7.6% of patients in the trastuzumab duocarmazine group had ILD/pneumonitis (grade ≥3: 2.4%). Altogether, there were 6 deaths reported with trastuzumab duocarmazine; of these, 4 were treatment-related (pneumonitis, n=2; respiratory failure, n=1; pneumonia, n=1).
A novel ADC composed of the backbone antibody trastuzumab (T) site-specifically conjugated to a derivative of the potent anthracycline (PNU) has recently been developed to evoke antitumor immunostimulatory effects.61 T-PNU enhances adaptive antitumor activity by reshaping the transcriptional and immunogenomic profile within the tumor microenvironment to provide long-lasting antitumor immunity and immune protection from subsequent tumor challenges.61,62 Studies have shown that due to these strong antitumor properties, T-PNU can overcome resistance in a breast cancer model unresponsive to trastuzumab and trastuzumab emtansine.61,63
In addition, multiple preclinical and early clinical studies are currently evaluating the receptor tyrosine kinase-like orphan receptor 1 (ROR1) as a targetable antigen to treat various tumors.64–66 ROR1 is a surface transmembrane receptor tyrosine kinase highly expressed in a number of hematological malignancies and solid tumors, with low expression in normal tissues. Results of a preclinical study demonstrated that NBE-002, a next-generation ADC that conjugates a humanized anti-ROR1 monoclonal antibody to a derivative of the highly potent anthracycline PNU-159682, is a highly effective and promising targeted therapy for the treatment of ROR1-positive TNBC.65 NBE-002 is currently evaluated in the first-in-human, open-label, multi-center, phase I/II NBE-002-01 study in adult patients with advanced solid tumors.66 The primary endpoint of the study is to assess the safety and tolerability of NBE-002, establish the recommended dose and evaluate its antitumor activity.
Mirvetuximab soravtansine is an anti-folate receptor alfa (FRα) ADC that is under investigation in ovarian cancer. Early phase trials have shown signs of its clinical activities in patients with platinum-resistant ovarian cancer and FRα-positivity assessed on immunohistochemistry (defined as ≥25% tumor cells with at least 2+ staining intensity).67 A subsequent phase III trial (FORWARD I) comparing mirvetuximab soravtansine versus chemotherapy of physician’s choice failed to demonstrate an improvement in PFS (4.1 months vs 4.4 months; HR=0.98) in patients with platinum-resistant ovarian cancer and medium or high FRα expression (50−74% and ≥75% representing medium and high expression, respectively).68 To evaluate the potential efficacy of mirvetuximab soravtansine in a better-selected population, the confirmatory phase III MIRASOL trial comparing mirvetuximab soravtansine monotherapy with investigator’s choice chemotherapy is ongoing in patients with high FRα expression (defined as ≥75% of cells with at least a score 2-staining intensity) and results are awaited.60,69
Safety concerns with ADCs
Tumor specificity and expression level are important considerations for the target antigen when developing an ADC.16 In an ideal world, the ADC agent would target lineage-specific antigens, highly expressed on target tumor cells and minimally expressed or absent on normal cells.70 In reality, nearly all target proteins are expressed to varying degrees on normal cells, leading to on-target toxicity with ADCs. Unlike many hematological malignancies, lineage-specificity does not apply to solid tumors. Indeed, off-target toxicity is thought to have been a major cause of dose-limiting toxicity for first-generation ADCs during clinical development in solid tumors.71,72 Furthermore, the internalization capacity of ADCs is closely correlated with high expression of surface antigen, which is why second-generation ADCs with microtubule inhibitor payloads such as maytansinoids do not demonstrate cytotoxic activity on cells with low antigenic expression because they preferentially kill dividing versus quiescent cells.73 Second generation ADCs such as trastuzumab deruxtecan were developed with a cleavable linker able to release part of the payload to the tumor microenvironment, affecting non-target antigen overexpressing cells. However, this mechanism of target-independent uptake of ADCs into normal cells, or bystander killing effect, thought to be a major component of off-target toxicity, remains poorly understood. Still, off-target effects, including hematologic, hepatic, neurologic and ophthalmic events, often lead to dose-limiting toxicities with current ADCs.74 For example, grade 3 and grade 4 anemia, neutropenia and peripheral neuropathy are consistently reported for MMAE ADCs, whereas thrombocytopenia and hepatic toxicity are commonly reported for emtansine-containing ADCs. Other AEs of particular interest in ADC clinical development include ILD with trastuzumab deruxtecan and the risk of hepatotoxicity with trastuzumab emtansine.27 Although the DESTINY-Lung01 trial reported high rates of ILD/pneumonitis, most cases were of low grade and manageable (18/24 cases grade 1−2, 4 cases grade 3 and 2 cases grade 5).45 However, ILD remains an important identified risk with trastuzumab deruxtecan treatment. Trastuzumab emtansine-based therapy, whether given as monotherapy or in combination with pertuzumab, increases the risk of all-grade and high-grade alanine transaminase (ALT)/aspartate aminotransferase (AST) elevations in breast cancer patients, and thus increases the risk of serious liver injury.74 Off-target toxicity is a major drawback with second-generation ADCs and several new approaches are being evaluated to potentially decrease these toxic effects, such as fractional dosing and the use of biomarkers to select suitable patients for combination therapy.75
Outlook on emerging combinations with ADCs
To further enhance treatment efficacy in patients with solid tumors, several ongoing trials are investigating ADCs in combination with other therapies, such as ICIs, targeted agents and chemotherapy. Notably, studies showed that ADCs may activate a strong antitumor immune response and can thus be effectively combined with ICIs.76–78
Trastuzumab emtansine in various combinations is currently being evaluated in patients with breast cancer, with some promising results. For example, when combined with neratinib, a tyrosine kinase inhibitor (TKI), trastuzumab emtansine demonstrated efficacy and safety in patients with metastatic HER2-positive breast cancer in a multicenter phase I study.79 This combination has been further assessed in a phase II trial. Furthermore, trastuzumab emtansine plus tucatinib, a HER2-specific TKI, demonstrated preliminary antitumor activity with acceptable toxicity in heavily pretreated patients with ERBB2/HER2-positive metastatic breast cancer with and without brain metastases.80 Although the addition of atezolizumab to trastuzumab emtansine did not improve PFS outcomes and was associated with adverse safety profile,81 this combination is further studied in a subpopulation of patients with PD-L1-positive, HER2-positive advanced breast cancer.82
Trastuzumab deruxtecan is also being assessed in combination with other agents in different tumor types in several ongoing clinical trials. Very recently, results were presented from the phase Ib, two-part, open-label DS8201-A-U105 study investigating trastuzumab deruxtecan with nivolumab in patients with HER2-expressing UC.83 In the primary analysis, trastuzumab deruxtecan plus nivolumab showed antitumor activity in patients with high HER2-expressing tumors, with safety consistent with previous reports on trastuzumab deruxtecan and nivolumab monotherapy. Furthermore, based on prior research showing that the combination of trastuzumab deruxtecan plus pertuzumab may be superior to trastuzumab deruxtecan alone in patients with HER2-positive metastatic breast cancer, this combination is now being evaluated in the open-label, phase III DESTINY-Breast09 trial.84 Adult patients (planned n=1,134) enrolled in the study will be randomized 1:1:1 to receive either trastuzumab deruxtecan plus placebo, trastuzumab deruxtecan plus pertuzumab or SOC treatment. The primary endpoint of the study is PFS per blinded independent central review (BICR). Another study on trastuzumab deruxtecan is the ongoing phase Ib/II DESTINY-Breast07 trial in patients HER2-positive metastatic breast cancer in combination with other anticancer agents, such as durvalumab, paclitaxel, pertuzumab or tucatinib.85,86 In addition, the phase Ib/II DESTINY-Breast08 study aimed to investigate trastuzumab deruxtecan in patients with HER2-low advanced or metastatic breast cancer in combination with durvalumab, paclitaxel, capivasertib, anastrozole, fulvestrant or capecitabine.87 Trastuzumab deruxtecan plus anastrozole is further assessed in a phase II study for the treatment of early-stage HER2-low, HR-positive breast cancer.88
At the 2022 ASCO Genitourinary Cancers Symposium, data were reported from cohort 3 of the phase II TROPHY-U-01 trial, demonstrating that treatment with sacituzumab govitecan plus pembrolizumab led to encouraging antitumor activity, with a manageable safety profile and no new safety signals in ICI-naïve patients with mUC.89 The addition of pembrolizumab to sacituzumab govitecan is also being evaluated in PD-L1-negative mTNBC in the randomized phase II Saci-IO TNBC trial.90 Sacituzumab govitecan plus pembrolizumab will be further investigated as frontline therapy in a global phase III study versus standard of care pembrolizumab plus chemotherapy in first-line patients with locally advanced or mTNBC.91 Data also showed a synthetic lethality between sacituzumab govitecan and talazoparib, a PARP inhibitor. This combination is under investigation in mTNBC in a phase Ib/II study, which is currently recruiting patients.92
Further ongoing trials evaluating ADCs in various combinations across several tumor types are presented in Table S1 in the Supplementary Appendix.

Conclusions
While recent advances in our understanding of the importance of the targeting properties of an antibody and appropriate linker have improved the therapeutic index of ADCs, advancements in bioengineering techniques have also contributed to the development of several new bystander payloads. Indeed, the increasing number of new and accelerated drug approvals has contributed to the rapid market growth of ADCs in recent years. Second-generation ADCs to treat patients with advanced breast cancers are currently available in the US, Europe and Switzerland. Still, approval of several more ADCs for different indications is anticipated imminently in Europe, with US approvals already leading the way. Although there has been significant progress in the field of solid tumors with six ADCs currently approved in the US, achieving effective and nontoxic ADCs remains a major challenge. Improvements in ADC design and development that address current limitations together with an improved safety profile of ADCs will lead to more effective and less toxic ADCs in the foreseeable future.
CONFLICTS OF INTEREST
IC: Advisor/expert opinion: Astra Zeneca, MSD, GSK, Novartis; Travel grants: Tesaro; Institutional funding (Principal investigator for clinical trials): Bayer, MSD, Oasmia
Author Contributions
All authors contributed to and approved the final manuscript.