Introduction
Ovarian cancer (OC) is the 7th most common type of cancer in women worldwide and the 5th most common cause of cancer-related death in women in Western nations.1 Overall prognosis is poor, with a 5-year relative survival rate of 30.2% for advanced-stage disease.2 Delay in diagnosis is a clinically significant issue, with at least 70% of OC patients having International Federation of Gynecology and Obstetrics (FIGO) stage III or IV disease at diagnosis.3
Epithelial ovarian cancer (EOC) represents approximately 90% of all OC1 and comprised four histological subtypes (serous, mucinous, clear-cell, and endometrioid) based upon their distinct tissue architecture and morphology. High-grade serous ovarian cancer (HGSOC) is the most common EOC histotype.4 Different OC subtypes have different molecular characteristics, which in turn influence current standard treatment.5
TREATMENT OF ADVANCED EOC WITH PARP INHIBITORS
Optimal cytoreductive surgery to no residual disease followed by platinum-based chemotherapy remains the gold-standard treatment for newly diagnosed EOC.6,7 For patients with high-risk disease (residual tumor after primary debulking surgery or stage IV), the addition of the anti-angiogenic agent bevacizumab has improved patients’ outcome and became part of the standard of care.6 Ultimately, even with an excellent initial response, more than 80% of patients with advanced-stage EOC will experience disease recurrence.7
Poly (ADP ribose) polymerase inhibitors (PARPi) are newer drugs that have transformed the treatment paradigm of EOC.8 PARPi have shown to improve patients outcome in different settings: maintenance treatment in first-line,9–11 maintenance in platinum-sensitive recurrence12–14 or as a new line of treatment.15,16 Thus, most patients with EOC will receive a PARPi treatment at some point during their disease course. Despite these promising results, many patients will inevitably develop primary or acquired resistance to PARPi and as of today, no approved agents are available to effectively treat patients after progression to these targeted agents. Different mechanisms of PARPi resistance have been proposed, some of them similar to the ones responsible for platinum resistance.17,18 Understanding these mechanisms and developing new therapies to overcome them is a key priority for current clinical and translational research in ovarian cancer.
MECHANISMS OF PARPi RESISTANCE
Homologous recombination (HR) has been identified as one of the main pathways involved in repairing double-strand (DSB) DNA breaks resulting from DNA damage.19 In tumor cells with defective HR (homologous recombination deficiency, HRD) repair pathways, PARP modulates repair of DNA single-strand (SBB) breaks and thus enables cell survival.17–19 In tumor cells with HRD, PARPi trap PARP on DNA to block SSB repair, promoting the accumulation of fatal DSB and DNA damage and triggering cell death through the well-described mechanism of synthetic lethality.19–21
Approximately 50% of all EOCs are characterized by HRD, mainly due to genomic alterations in HR genes, including but not limited to BRCA1/2, and this correlates with a higher sensitivity to platinum-based chemotherapy and PARPi.22 Nevertheless, PARPi are less effective in cancer patients with proficient HR tumors.10,11,13 Thus, HRD and BRCA status are now established biomarkers for predicting responsiveness to both platinum chemotherapy and PARPi. Different HRD tests have been developed (e.g., tumor mutations in HR genes, genomic scars assessment [myChoice Myriad HRD test, FoundationFocus CDxBRCA, Foundation Medicin] or functional assays) to predict the magnitude of benefit from PARPi. However, these currently available tests have a low negative predictive value and more reliable assays are urgently needed.23
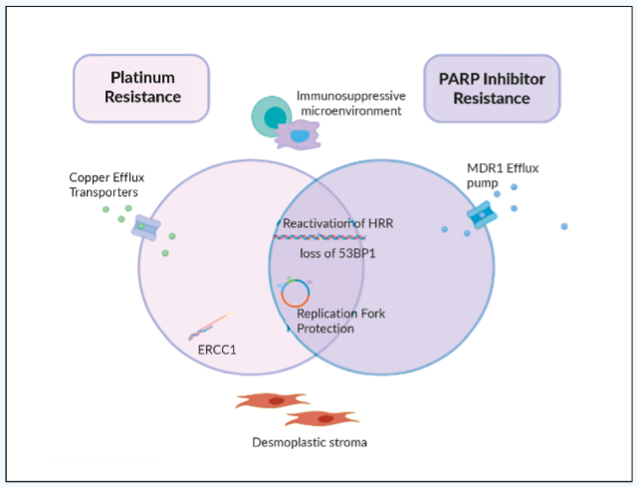
Different mechanisms sustaining PARPi resistance have been described. One of the major mechanisms of PARPi and platinum-based chemotherapy resistance is the restoration of HR capability re-enabling cancer cells to repair DSBs, thereby reducing genomic instability and sensitivity to PARPi (Figure 1).17,18 Restoration of HR might occur due to epigenetic events leading to re-expression of HR genes or to secondary mutations that re-established gene function (reversion mutations) or upregulation of the wild type allele.24–26 For example, somatic BRCA reversion mutations that restore BRCA function have been reported and is now a well-established mechanism of PARPi resistance.24–26 Other mechanisms include reactivation of HR due to loss of DDR p53binding protein [53BP1], involved in the error-prone non-homologous end-joining repair,27 and stabilization of replication fork.

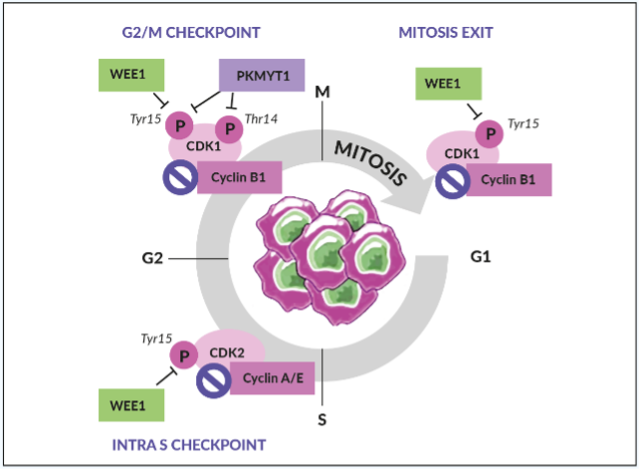
Another possible mechanisms of resistance are the activation of other pathways (PI3K/AKT/mTOR, RAS/RAF/MET, VEGFR) able to activate or restore HR and the increased dependence on other DNA repair mechanisms or cell cycle regulators.28,29 The cell cycle checkpoints (e.g. ataxia telangiectasia mutated protein [ATM], the ataxia telangiectasia and Rad3-related protein [ATR], p53, WEE1, checkpoint kinase 1 and 2 [CHK1 and CHK2]) coordinate the arrest of the cell cycle to repair the DNA damages and promote a high-fidelity cell replication.30
Notably, about 95% of HGSOC harbor mutant TP53, a tumor suppressor gene involved in regulating the first G1/S cell cycle checkpoint. Tumor cells deficient in the first checkpoint are more dependent on the G2/M checkpoint and the WEE1 pathway for DNA repair.
WEE1 KINASES AND INHIBITORS
Mechanisms of action
WEE1 is an inhibitory kinase that plays an important role in cell cycle regulation and DDR, particularly as a gatekeeper of the G2/M stage of the cell cycle (Figure 2).31 WEE1 also regulates DNA replication and chromatin integrity during the S phase. In response to DNA damage, WEE1 directly inhibits the downstream cyclin-dependent kinase (CDK) 1 and CDK2 protein complexes, thus preventing mitotic entry and replication of cells with altered DNA.
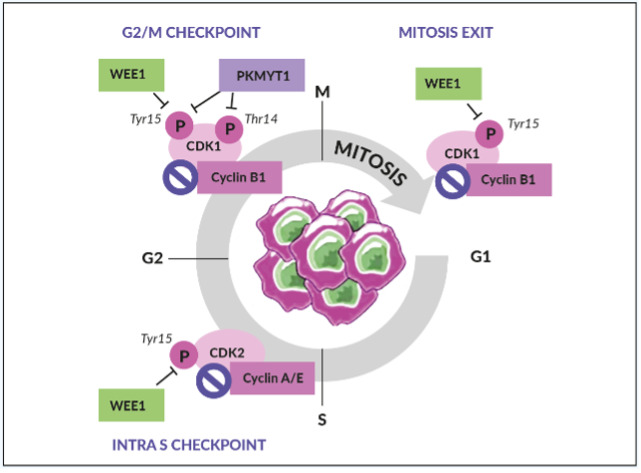
WEE1, therefore, acts as a tumor suppressor in healthy, non-malignant cells.31 Aberrant WEE1 gene expression has been observed in various human cancers. For example, WEE1 gene overexpression has been found in 35% of breast cancers, especially in human epidermal growth factor receptor 2 (HER2)-positive subtypes, and 92% of OCs.32,33 In contrast, WEE1 gene suppression was found in human colon cancer cell lines analyzed using complementary DNA (cDNA) array techniques.34 WEE1 inhibition removes the brakes on cell cycle progression and increases DNA damage, which leads to catastrophic mitosis and tumor cell death. Together, these observations provided the rationale for investigating WEE1 inhibition as a viable therapeutic anticancer strategy.35–37
OVERCOMING PARPi AND PLATINUM RESISTANCE WITH WEE1 INHIBITORS
Preclinical and clinical evidence for combining WEE1 inhibitors with chemotherapy
Impacting both DDR and cell cycle progression, WEE1 inhibition may increase cell death in response to chemotherapy and ionizing radiation. A proof-of-concept study in pancreatic cancer cells showed that WEE1 inhibition sensitized HR-proficient (BRCA2 wild-type) cancers but not HRD-positive (BRCA2 mutant) cancers to gemcitabine-based chemoradiation.38 This preclinical study suggests that WEE1 inhibitors are involved in the inhibition of HR repair as well as abrogation of the G2 checkpoint.38
Adavosertib, a first-in-class selective small-molecule inhibitor of WEE, prevents WEE1 from phoPreclinical and clinical evidence sphorylating the Tyr15 residue of CDK1, thus activating the cyclin B1–CDK1 complex to enable cells to enter mitosis.31 Evasion of the G2 phase forces cells harboring DNA lesions to progress into mitosis, inducing premature mitotic exit and apoptosis.31,37 Early-phase clinical trials in several adult solid tumors have shown that adavosertib has promising antitumor activity as monotherapy or in combination with chemotherapy drugs such as 5-fluorouracil (5-FU), gemcitabine, carboplatin and cisplatin.39–41
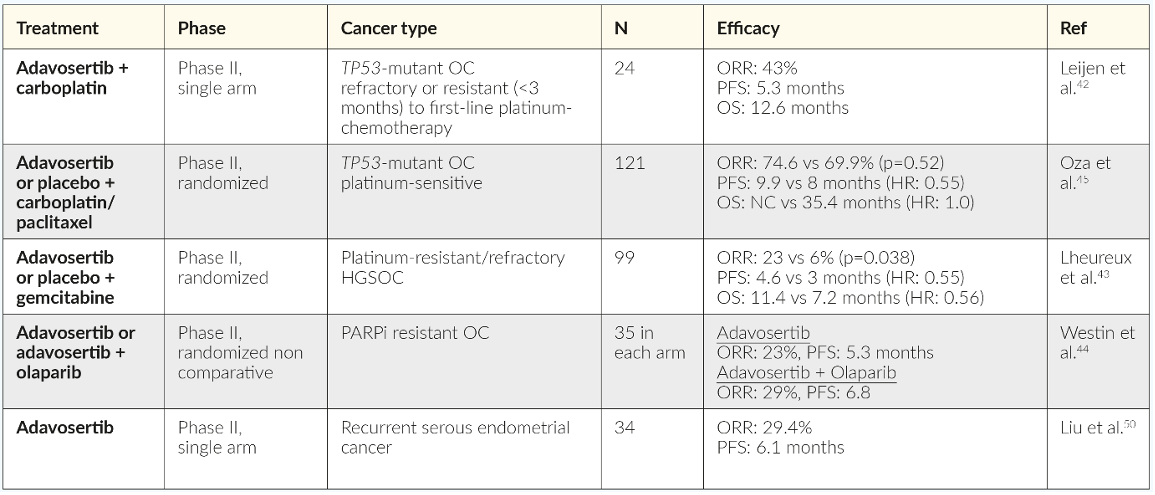
Several clinical studies have investigated the strategy of synthetic lethality by exploring simultaneous inhibition of multiple DNA repair mechanisms, for example, dual inhibition of WEE1 and PARP in combination with other DNA-damaging agents in patients with tumors harboring BRCA mutations or the combination of a WEE1 inhibitor and chemotherapy.42–45 Notably, clinical evidence for sensitization of platinum-resistant EOC cells to platinum-based chemotherapy has been demonstrated with WEE1 inhibitors (Table 1). A phase II study reported by Leijen et al. (2016) provided clinical proof that the WEE1 inhibitor, adavosertib, could enhance the efficacy of carboplatin in patients with TP53-mutated EOC refractory or resistant (<3 months) to first-line platinum-based therapy.42 Another randomized phase II placebo-controlled study investigated the role of adding adavosertib to carboplatin and paclitaxel in patients with TP53-mutated platinum-sensitive recurrent EOC, showing an improvement in progression-free survival (PFS) (9.9 months vs 8.0 months, HR: 0.55 [95% CI: 0.32–0.95]).45 Furthermore, in a double-blind, randomized, placebo-controlled phase II study in patients with platinum-resistant or platinum-refractory advanced HGSOC, adavosertib combined with gemcitabine significantly improved PFS (4.6 months vs 3.0 months, HR 0.55 [95% CI: 0.35–0.90]) and overall survival (OS) (11.4 months vs 7.2 months, HR: 0.56, [95% CI: 0.35–0.91]) versus placebo.43 Notably, signs of activity of adavosertib plus gemcitabine were also observed in an exploratory cohort of rare histological subtypes, including low-grade serous or endometrioid, carcinosarcoma or clear cell OC.43 As expected, the combination of adavosertib and chemotherapy resulted in increased toxicity, mainly hematological that frequently required dose interruption or reduction.42,43,45

Preclinical and clinical evidence for combining WEE1 and PARP inhibitors to overcome PARPi resistance
Preclinical studies showed that the inhibition of WEE1 might reverse resistance to PARPi. In pancreatic cancer cells, the addition of a WEE1 inhibitor in HR-proficient tumors enabled maximal radiosensitization by PARPi.46 Therefore, these two agents may work in synergy to impair DNA replication at multiple levels.
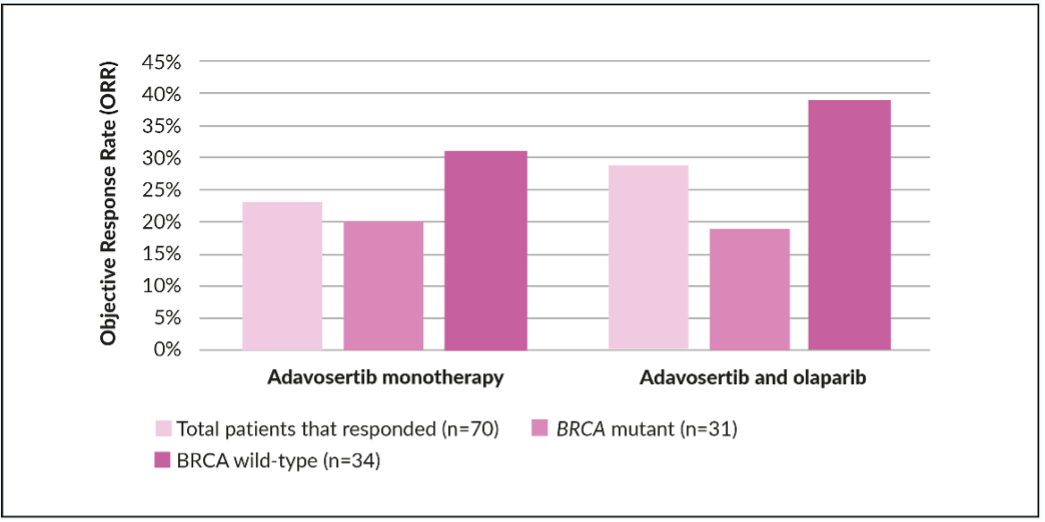
The benefit of dual inhibition of HR repair pathways in patients with advanced OC using a combination of WEE1 inhibitors and PARPi was recently examined in the EFFORT (EFFicacy off adavosertib in parp ResisTance) study (Table 1). EFFORT (clinical trial identifier: NCT03579316) is a randomized, two-arm, non-comparative phase II study that evaluated adavosertib with or without the PARPi olaparib in 80 women with PARPi-resistant OC.44 The overall response rate (ORR) was 23% for patients in the adavosertib monotherapy arm and 29% in the combination arm (Figure 3). The clinical benefit rate (objective response and stable disease >4 months) for patients receiving adavosertib alone or in combination with olaparib was 63% and 89%, respectively. Median PFS was 5.5 months (90% CI: 3.9−6.9) in the monotherapy arm and 6.9 months (90% CI: 4.3−8.3) in the combination arm.44 Efficacy of adavosertib as a single agent or in combination with olaparib was demonstrated regardless BRCA status. Among women with BRCA mutations, the ORR was 20% with adavosertib monotherapy (n=15) versus 19% with the combination (n=16) (Figure 3), with a median duration of response of 5.6 months and 6.4 months, respectively, and clinical benefit rate of 67% and 81%, respectively. In patients with BRCA wild-type, ORR was 31% with adavosertib (n=16) and 39% with adavosertib and olaparib (n=18) and the clinical benefit rate of 69% versus 94%. The median duration of response was 4.1 and 8.7 months, respectively.44

Patients in both monotherapy and combination treatment arms experienced treatment-associated adverse events, mainly hematological and gastro-intestinal. Despite these toxicities were usually manageable with supportive care, dose interruptions (72% monotherapy vs 88% combination arm), or dose reductions (51% vs 71%) were necessary.44 Further research is warranted to define the optimal dose and schedule of adavosertib in combination with PARPi, to improve tolerability and dose intensity and to maximize patient benefit. Fang et al. (2019) showed that adding a WEE1 inhibitor and PARPi in sequence minimizes toxicity in TP53-mutant and BRCA wild-type OC model while maintaining efficacy and avoid the onset of drug resistance.47 A phase I trial is ongoing (STAR trial, NCT04197713) to asses the sequential combination of adavosertib and olaparib in patient with advanced solid tumors with selected HR genes mutations and failure to previous treatment with a PARPi.
WEE1 inhibitor in other gynecological malignancies
The efficacy of the WEE1 inhibitor adavosertib has been also investigated for the treatment of advanced recurrent serous endometrial cancer following failure of platinum-based chemotherapy. Serous uterine cancer is a distinct histological subtype of endometrial cancer with, similarly to HGSOC, an high prevalence of TP53 mutations and high replication stress.48 A single-arm phase II study investigated the efficacy of adavosertib among 34 evaluable patients, with an ORR of 29.4% with a median duration of response of 9 months and a PFS of 6.1 months.49 Despite these results suggest a possible new therapeutic opportunity for patients with endometrial cancer, further research is needed to define predictive biomarkers and confirm these preliminary findings. An international phase II study is ongoing (ADAGIO, NCT04590248) to assess the efficacy of adavosertib in a larger population of women with serous uterine cancer progressed after at least one prior line of platinum-based chemotherapy.
Conclusions
As the primary G2/M cell-cycle checkpoint gatekeeper, the WEE1 pathway can be used to exploit intrinsic replication stress and dysfunctional checkpoint in tumor cells. Therefore, the rational combination of a WEE1 inhibitor to DNA-damaging chemoradiotherapies or PARPi may be a viable anticancer treatment strategy for various tumor types. Promising preclinical and early clinical trial results show that combined WEE1 inhibition enhances the effect of chemoradiotherapy or PARPi in patients with platinum- or PARP-resistant ovarian cancer regardless of BRCA status. One of the main limitations for the clinical development of this compound, both as single agent and in combination, is represented by the high rate of hematological toxicities, often requiring dose reduction or interruption, particularly in heavily pretreated population. However, the timing of combination therapy is important to obtain optimal treatment efficacy and improve tolerability. Studies determining the optimal regimen and schedule to combine DNA damaging agents and WEE1 inhibitors to treat advanced ovarian cancer are ongoing and the results will provide further insight on how better administered these agents. No biomarkers able to predict WEE1 efficacy and to support a better patient’s selection are currently available and further results from translational research are eagerly awaited. Moreover, other WEE1 inhibitors (IMP7068, Debio0123) are under development and will give further information into the potential role of these agents for the treatment of EOC and other solid tumor types.
Take-Home Messages
- It is possible to combine DNA damage response inhibitors, such as cell cycle checkpoint inhibitors, chemoradiation or PARP inhibitors, to enhance antitumor activity.
- More than 95% of patients with high-grade serous ovarian cancer harbor mutant TP53, suggesting that the G2/M checkpoint is a potential anticancer target.
- Several preclinical and clinical trials have investigated the efficacy of the WEE1 inhibitor adavosertib as a single agent or in combination with other DNA-damaging agents or PARP inhibitors.
- In patients with PARPi resistant EOC, WEE1 has shown early signs of activity when used as a single agent or in combination with olaparib, regardless of BRCA status, warranted further exploration.
CONFLICT OF INTEREST
Travel grants: Tesaro
External expert: AZ, GSK, Novartis
Institutional grants for clinical trials (PI): MSD, Bayer, Oasmia
Author Contributions
The author crafted and approved the final manuscript.