


Introduction
Clonal hematopoiesis of indeterminate potential (CHIP) refers to the presence of somatic mutations in known oncogenes in hematopoietic stem and progenitor cells (HSPCs) that were previously implicated in hematologic malignancies with a variant allele frequency (VAF) of at least 2%, but without the presence of hematologic cancer or other clonal diseases.1 Very small clones of clonal hematopoiesis are present in nearly all humans above the age of 40 years, whereby CHIP is age-dependent with a prevalence of about 10% in people aged around 70 years.2–6 Smoking and other genotoxic stressors have also been shown to increase the risk of clonal hematopoiesis.3 Chronic inflammation in tumor microenvironments is known to promote malignant transformation and cancer progression in general, although the underlying mechanisms that facilitate hematopoietic neoplasms in bone marrow are far from being completely understood.7 In addition to hematologic cancers, experimental evidence suggests that CHIP is a risk factor for all-cause mortality, thromboembolism, and cardiovascular diseases, and that it contributes to the development of atherosclerosis and cardiac dysfunction.8
Healthbook TIMES Oncology Hematology previously introduced this topic in 2020, with a description of the development of CHIP and its implications in cardiovascular diseases and cancers.9 Studies using advanced sequencing technologies might provide valuable insights into the interrelationships between clonal hematopoiesis, cancer, inflammation, and cardiovascular complications, as well as the clinical consequences of CHIP. This article summarizes the latest evidence from the most recent CHIP studies.
Clonal hematopoiesis is defined as recurrent mutations in known oncogenes in hematopoietic stem and progenitor cells (HSPCs) in the absence of overt hematologic malignancies.10 It is an age-related condition that is associated with increased risk of hematologic cancers, such as therapy-related myeloid malignancies, cardiovascular disease, thromboembolism, and all-cause mortality.
INSIGHTS FROM CHIP SURVEILLANCE STUDIES USING NEXT-GENERATION SEQUENCING TOOLS
Age-related clonal hematopoiesis
In the last few years, there has been rapid expansion in the use of next-generation sequencing (NGS)‐based approaches in cancer research and diagnostics.11 NGS studies have revealed that CHIP-associated mutations can be found in apparently healthy individuals with normal blood counts, and especially in older adults.1 Using whole exome sequencing, recent NGS studies estimated that hematopoietic clones are present in approximately 10% of individuals aged over 65 years, and in more than 20% of individuals aged over 90 years (Figure 1).2–5 Notably, the rate of CHIP progression to hematologic neoplasm appears to be the same as for other known clonal pre-malignant disorders, such as the transition of monoclonal gammopathy of undetermined significance (MGUS) to multiple myeloma, which has been estimated at 1% per year.1,8,12,13 It remains unknown why some individuals who harbor somatic mutations sustain clonal expansion of these mutations, or even progression to hematologic neoplasms, while others do not.14
_by_age.png)
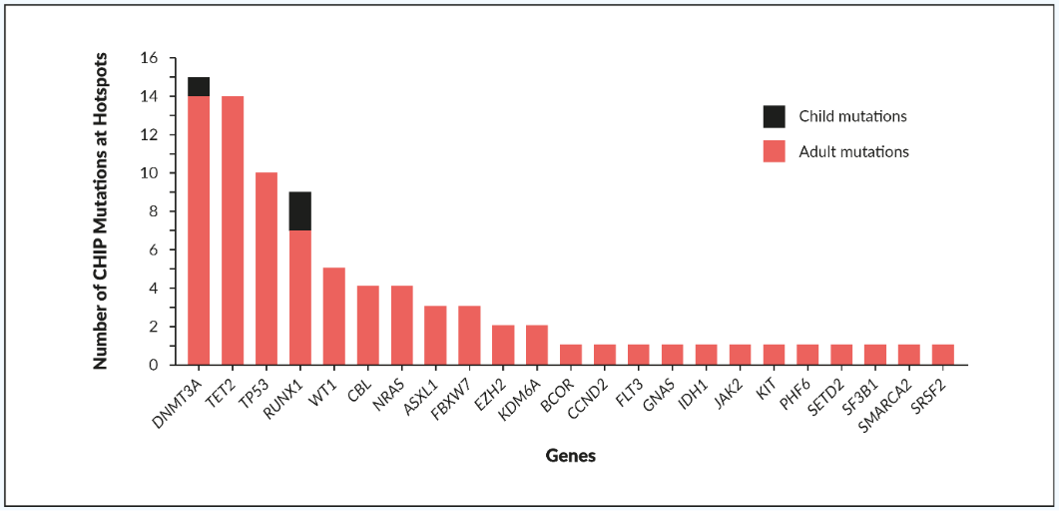
A recent literature review of NGS studies by Feusier et al. (2021) identified 20,141 protein-altering mutations in 7,430 patients with leukemia or other hematologic malignancies.15 Of these, they identified 434 significantly recurrent mutation hotspots across 85 genes, 364 of which occurred at loci that are confidently assessable for CHIP. Of 755 constituent mutations, 134 had been reported at diagnosis in at least three patients with hematologic malignancy.
Although CHIP was previously believed to be identifiable in adults only, 3 of 388 (0.77%) children in a cohort of 796 individuals without blood disorders were shown for the first time to harbor CHIP mutations at leukemic hotspots.15 Whether the rate of CHIP mutation in these children was higher than expected based on data from adult studies remains to be determined.
CHIP mutation hotspots and nonhotspots
Somatic mutations associated with CHIP are mainly seen for three genes that encode epigenetic regulators involved in the control of hematopoiesis: DNA methyltransferase 3α (DNMT3A), tet methylcytosine dioxygenase 2 (TET2), and additional sex combs like 1 (ASXL1).3,4,16 The significant recurrence of CHIP mutations in hematologic malignancies might provide prognostic information to determine the risk of developing blood cancers.
Feusier et al. (2021) reported clones with CHIP mutations that were identical to those identified in significant hematologic malignancy hotspots in 83 of 4,530 (1.83%) apparently healthy individuals without cancer.15 In the noncancer individuals, CHIP mutations were most frequently identified in the DNMT3A, TET2, and tumor protein 53 (TP53) genes (Figure 2). A further 183 individuals without cancer harbored nonhotspot CHIP mutations in genes with known hematopoietic function, most frequently as TET2 and DNMT3A.15 Most CHIP patients had only a single clonal mutation in one of the genes that regulates hematopoiesis, which suggests that most of the CHIP mutations detected, at both hotspot and nonhotspot loci, were probably drivers of the clonal expansion. Which specific subgroups should be screened for CHIP to benefit from early interventions is an area of intense research.
_detected_at_hotspots_in_noncancer_i.png)
CHIP surveillance in routine cancer care
Evidence from the literature suggests that tumorigenesis is a multi-step process that depends on (among others) gene mutations and chromosomal alterations.17–19 A recent framework introduced by Gao et al. (2021) explored the potential to enable comprehensive clonal hemopoiesis surveillance programs to be readily integrated into routine cancer care.20 The results of their study suggested that in the routine surveillance of patients at risk of hematologic neoplasms, mosaic chromosome alterations should also be considered together with gene mutations.
Cell-free DNA (cfDNA) testing
Cell-free circulating tumor DNA (ctDNA) is a fast-emerging liquid biopsy biomarker for the characterization of genetic aberrations in cancer.11 For the majority of cancers it holds true that the earlier the detection, the higher the chance of a potential cure. Phallen et al. (2017) detected ctDNA using an NGS assay in 62% of patients with diagnosed stage I–II breast, lung, ovarian, or colorectal cancers.21 Thus, ctDNA might become an attractive method for cancer screening in the future, to perhaps reduce other screening analyses like colonoscopy or mammography, or as a follow-up-examination also in solid tumors. Indeed, in several hematologic malignancies, it is already an established method. One challenge for implementing ctDNA in clinical practice is the differentiation of a cancer signal from the background normal biological variation within an individual. Notably, more than 80% of cell-free DNA (cfDNA) in healthy individuals arises from hematopoietic cells, so cfDNA analysis can be used for genetic profiling of hematologic cancers.22 CHIP-associated mutations, aging, and inflammatory factors can create “biological noise” by increasing the levels of normal cfDNA. The subset of cfDNA carrying specific genetic aberrations from CHIP can lead to a high false-positive rate and potential misdiagnosis, and thus to inappropriate treatment. Indeed, Liu et al. (2019) found at least one nonsynonymous mutation in 60% of cfDNA samples from healthy individuals.23
Cell-free DNA screening is increasingly used in advanced prostate cancer to determine eligibility for poly (ADP-ribose) polymerase (PARP) inhibitor treatments. Based on DNA repair gene status, several PARP inhibitors have become commercially available, including olaparib and rucaparib.24 CHIP mutations that involve DNA repair genes (e.g., BRCA, ATM) present biological confounding factors that can produce false-positive results in a large proportion of patients. Jensen et al. (2021) reported that 10% of men with advanced prostate cancer had CHIP mutations in DNA repair genes that interfered with plasma cfDNA testing. Since inaccuracies with commercial plasma cfDNA testing kits have come to light, cfDNA testing of DNA repair gene status should include a whole-blood control to distinguish CHIP from prostate-cancer-related variants.
CLINICAL SIGNIFICANCE OF CHIP
Association between clonal hematopoiesis, atherothrombotic disorders, and cardiovascular conditions
Somatic mutation-driven clonal expansion of mutant hematopoietic cells without evident disease has many far-reaching clinical consequences that have not been considered previously. In preclinical and clinical studies, CHIP was associated with nearly a doubling of coronary heart disease risk and accelerated atherosclerosis.25 Jaiswal and Libby (2020) recently reviewed the association between CHIP, inflammation, and numerous other age-related diseases, such as cancer and cardiovascular disease.26 Although they confirmed that there are no treatments currently available to lower the risk of cardiovascular disease in individuals with CHIP, they suggested that blockade of inflammatory molecules might represent a strategy to mitigate the effects of CHIP. Moreover, they recommended that healthy individuals who are incidentally found to have CHIP should undergo evaluation for lifestyle modifications, to reduce the risk of cardiovascular disease.
Veninga et al. (2020) considered the potential relationships between the driver genes associated with clonal hematopoiesis and platelet function related to both (athero)thrombotic and hemostatic disorders.27 A literature search identified seven relevant CHIP genes associated with elevated platelet counts and the associated thrombotic risk: ABCB6, ASXL1, DNMT3A, GATA1, JAK2, SF3B1, and SH2B3 (Figure 3). Further studies are needed to determine the functional status of platelets in the context of CHIP mutations and their contribution to the risk of thrombosis.
_that_affect_platel.png)
The role of CHIP in transplant complications
For patients undergoing either allogeneic hematopoietic stem cell transplantation (allo-HSCT) or autologous stem cell transplantation (ASCT), pre-existing clonal hematopoiesis is associated with adverse outcomes.10,28 Gibson et al. (2017) reported on a cohort of lymphoma patients who underwent ASCT, and showed decreased overall survival (OS) in 120 patients with pre-existing CHIP, compared to 272 patients without pre-existing CHIP (10-year OS, 30.4% vs 60.9%; p<0.001). This included increased risk of death from therapy-related myeloid neoplasm (TMNs) (i.e., myelodysplastic syndrome, [MDS] acute myeloid leukemia [AML]) and cardiovascular disease.28
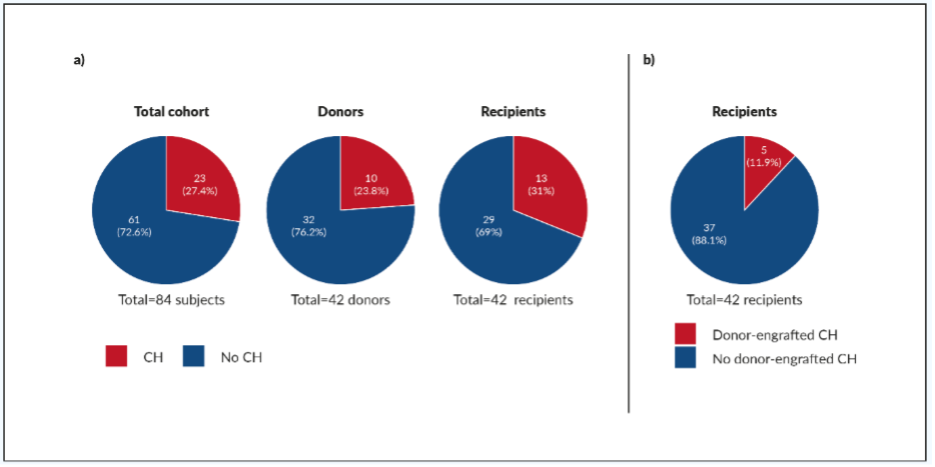
During allogeneic transplantation, a small proportion of hematopoietic stem cells are transferred to the recipient from the donor. In the recipients, donor-engrafted clonal hematopoiesis can increase proliferation within an inflammatory microenvironment and drive clonal expansion. Donor-engrafted clonal hematopoiesis has been reported to be common in allo-HSCT recipients.10,29 Boettcher et al. (2020) evaluated the effects of pre-existing clonal hematopoiesis in long-term survivors of allo-HSCT and their respective donors.10 The clonal hematopoietic profiles of 42 allo-HSCT donor–recipient pairs were analyzed by NGS, with a median follow-up of 16 years. At least one somatic mutation was detected in 10 of 42 (23.8%) donors and 13 of 42 (31.0%) recipients (Figure 4a). Furthermore, donor-engrafted clonal hematopoiesis was found for 5 of 42 (11.9%) transplantations; i.e., in 5 of 10 (50.0%) donors, and 5 of 13 (38.5%) recipients with clonal hematopoiesis (Figure 4b). One of the 5 donor-engrafted clonal hematopoiesis cases progressed into MDS in both donor and recipient, and 4 of the 5 cases showed clonal size increase. These studies provide further insights into the potential impact of clonal hematopoiesis, and prompt further research into how premalignant clonal hemopoiesis progresses to myeloid malignancy.
_in_donors_and_recipients_of_allogeneic_hematopoietic_stem_cell_t.png)
Donor-derived leukemia (DDL) is a relatively rare, but severe, complication of HSCT, and it accounts for approximately 5% of leukemia relapses.16 Patients typically show poor prognosis, with a median time to death after diagnosis of 10.6 months. Whereas traditional relapses occur shortly after HSCT, DDL often shows a longer latency (approximately 26 months). This indicates that significant changes occur that drive tumorigenesis. Several mechanisms might have a role in CHIP and/or DDL pathogenesis, such as unexplained cytopenias and the inflammatory response that can occur due to chronic graft-versus-host disease. Burns et al. (2020) postulated that the mechanisms underlying cGVHD or myelodysplastic syndrome might facilitate the expansion of CHIP clones, the acquisition of new mutations, or the changes in hematopoiesis that promote DDL (Figure 5).
_and_myelodysplastic_syndromes_(mds)_might_represent_.png)
Future directions
Although recent studies have provided insights into the impact of CHIP mutations on the development of hematologic neoplasms, the pathogenesis process has yet to be fully characterized.14–16 Improved understanding of the biology of CHIP mutations will help to provide early and targeted interventions for cancer patients. Therefore, further studies are required to better understand the pathogenic mechanisms that link age-related inflammatory disorders such as cardiovascular disease and (athero) thrombosis with hematologic malignancy. Genomic profiling by NGS and targeted panel sequencing using cfDNA are increasingly being used for tumor surveillance and CHIP monitoring in clinical studies.15,23,24 The introduction of advanced sequencing technologies into routine clinical practice in conjunction with improved interpretation of results will ultimately enable tailor-made interventions for patients with hematologic neoplasms, to provide preventive care.20 By conducting clinical trials and establishing guidelines to manage subgroups of patients with CHIP mutations, a new level of care can be provided to help mitigate the risk factors associated with CHIP.
Conclusions
-
There is increasing evidence that CHIP (which affects around 10% of the general population in their 7th decade of life in an age-dependent manner) can adversely affect human health.2–5
-
Next-generation sequencing (NGS) applications have revealed connections between CHIP and medical conditions (e.g., cancer, cardiovascular diseases).15
-
For the fast-emerging cfDNA-based screening in routine clinical practice, allowing for CHIP is important to avoid false-positive results.23,24
-
Further studies into the clinical and biological implications of CHIP are warranted.
CONFLICT OF INTEREST
The author declares that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
The author crafted and approved the final manuscript.