
The early days of immunotherapy
The first documented case of immunotherapy was performed by Dr William Coley, a bone surgeon and oncologist, in the 1890s in New York. He documented a correlation between accidental infections and spontaneous tumor regressions in cancer patients.2 Based on these retrospective observations, he successfully treated a 35-year old man with a terminal case of neck cancer with repeated intra-tumoral injections of live bacteria. Although, at that time, the function of the immune system was barely recognized, experts today agree that the effect induced by Dr Coley should be considered as an early form of immunotherapy. Despite these promising results, immunotherapeutical approaches were overtaken by developments in radio- and chemotherapy in the first half of the 20th century. The ground-breaking work of Dr Old, who sought to treat cancer by improving the body’s natural defences against it, was therefore widely considered odd. In the 1970s, Dr Old successfully performed intravesical injections of Bacillus Calmette-Guérin (BCG), a live vaccine against tuberculosis, in patients with bladder cancer. This is considered to be the first direct demonstration that the immune system can be used against cancer, and today, BCG is approved for the treatment of non-invasive bladder cancer.3,4
T cells recognize and attack cancer
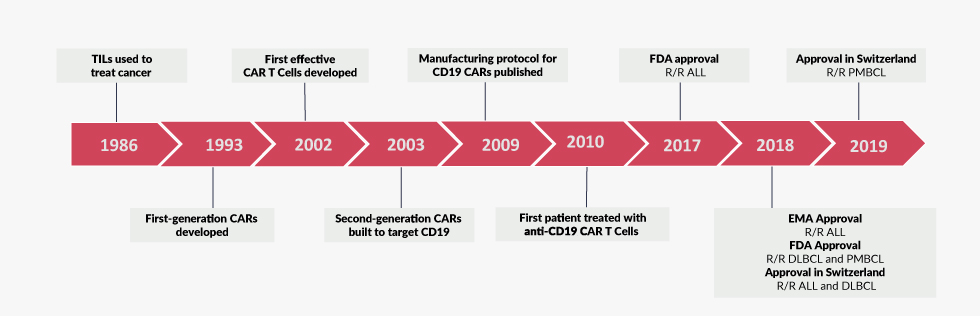
Starting from the 1950s, many of the fundamental immunological mechanisms were discovered, which have become textbook knowledge today. For instance, the thymus was recognized as an immunological organ in which T cells develop.5 The identification of protective immunity conveyed by lymphocytes against sarcoma cells in an experimental setting in 1960 added an unexpected aspect to the repertoire of T cell functions.6 In human patients, T cells were observed to accumulate in cancer tissue, further indicating a surveillance function of lymphocytes against malignantly transformed host cells. In the 1980s, Dr Steven Rosenberg from the National Institute of Health (NIH) pioneered the field of adoptive T cell transfer as he extracted tumor-infiltrating lymphocytes (TILs) from resected melanomas, expanded them in vitro and achieved durable remissions upon re-administration into patients (Figure 1).7,8 Despite the curative potential of adoptively transferred TILs,8 the complex manufacturing process as well as the limited efficacy prevented a widespread clinical implementation of this technique. Functionally, TIL-mediated recognition of antigens relies on an endogenous T cell receptor, which exhibits moderate affinity to cancer antigens and requires the presence of major histocompatibility complex (MHC) molecules.9 Much later, tumor immune evasion mechanisms, such as the down-regulation of MHC molecules or the up-regulation of negative immune regulators on tumor cells were demonstrated to critically limit the anti-tumor functions of unmodified T-cells.10

With the knowledge gained from studying the human immunodeficiency virus (HIV), replication-defective retroviruses were developed as a novel method for gene transfer by the late 1980s. With this technology, foreign genes could be stably introduced into eukaryotic cells for the first time.11,12 Additionally, with the development of polymerase-chain-reaction (PCR), DNA sequencing and molecular cloning techniques, the generation of artificial/synthetic genes became possible. With these new possibilities, the idea of combining the specificity of antibodies with the effector strength of T cells, using an intentionally designed and synthetically produced chimeric gene, was born. Kuwana et al., envisioned that the expression of an antibody-like molecule on the surface of T cells could be used to re-direct them against target cells in a non-MHC restricted manner.13 The design was subsequently refined by Eshhar et al.14 Albeit major differences exist in the design of the artificial gene in these pioneering works in comparison to modern CAR T cells, Eshhar’s modified cells were able to kill antigen-expressing target cells in vitro and thus provided evidence for the feasibility of targeted redirection of T cells for the first time. Notably, these cells did not contain an intrinsic activation domain and depended on integration of the CAR into the endogenous TCR complex for signaling. In 1991, three separate reports introduced the intracellular CD3ζ chain as the activating domain in different chimeric constructs, which facilitated the CAR design and drastically increased the activation potential.15–17
From bench to bedside
The current design of CARs is derived from these reports and consists of three different modules: an extracellular antigen-binding domain (1) that is linked via a stalk and transmembrane part (2) to an intracellular signalling domain (3). In all clinically relevant CAR constructs, the extracellular antigen-binding domain is formed by an antibody-like molecule, the so-called single-chain fragment variable (scFv). The design of the transmembrane domain differs and the length of the stalk must be optimized empirically for the respective antigen of choice. The diverging biological properties of different CAR T cells mainly result from the target antigen and the composition of the intracellular signalling domain. A common feature of all CAR T cell constructs is the incorporation of a CD3ζ stimulation domain, which mediates signal transduction from the T cell receptor (TCR) to the intracellular compartment in normal T cells.18 CAR T cells which only incorporated a CD3ζ chain in their signalling domain (first-generation CARs), demonstrated anti-cancer activity in vitro but failed to persist and to induce long-term remissions in clinical trials despite exogenous application of Interleukin-2.19
To overcome these limitations, CARs have been gradually refined. Importantly, the addition of co-stimulatory moieties in the signalling domain resulted in more robust and persistent CAR T cell activation. In fact, different co-stimulatory domains were introduced that imitate the activation mechanism of normal T cells, which is generally considered a three-step process. The first signal is the ligation of the TCR to an antigen-MHC complex on the surface of antigen-presenting cells. The second signal is mediated by ligation of a co-stimulatory molecule such as CD28 or CD137 (4-1BB) to their cognate ligands. These signals induce the synthesis of cytokines such as interleukin (IL)-2, which are essential for T cell differentiation and survival (signal three).
In seminal studies, Dr Michel Sadelain, Dr Hinrich Abken, Dr Weir and Dr Roberts independently designed second-generation CAR T cells which incorporated a co-stimulatory CD28 chain in the signalling domain.20,21 These cells conferred proliferative advantage over first generation CAR T cells as well as an increased release of immunostimulatory cytokines, while maintaining their cytolytic potential. Increased biological potency of anti-CD19 second-generation CD28-CD3ζ CAR T cells compared to their first-generation counterparts became evident in pre-clinical models of acute lymphoblastic leukemia (ALL).22 With the potential of expansion in vivo, the foundation for the use of CAR T cells as “living drugs” was laid. Since then, several other co-stimulatory signals have been integrated into CARs including CD134/OX40, CD137/4-1BB and CD27.18 As mentioned before, the biological properties (i.e. cytokine release, T cell expansion and persistence) relate from the respective co-stimulatory domain. Finally, multiple co-stimulatory domains have been combined into a single construct (third-generation CARs). However, a clinical advantage of these constructs is yet to be shown.
Building on the promising pre-clinical data, first-in-human CAR T cell trials were initiated. The two initial studies, however, failed due to different reasons. CAR T cells targeting the ovarian cancer-associated antigen folate receptor alpha (FRα) were evaluated for the treatment of patients with metastatic ovarian cancer.23 Despite the manageable safety profile, responses were limited and not durable. In contrast, the infusion of CAR T cells targeting the carboxy-anhydrase-IX (CAIX) in patients with metastatic renal cell carcinoma (RCC) resulted in severe liver toxicities leading to treatment discontinuation in 2 out of 3 patients.24 Therefore, it became evident that the choice of target antigen was of paramount importance for clinical effectivity of CAR T cell therapy.
In 2003, Dr Sadelain’s group introduced CAR T cells targeting CD19, an antigen expressed by healthy and malignant B cells and thus set the stage for a revolutionary era in the treatment of hematological malignancies.25 On the basis of convincing pre-clinical data, large academic cancer institutes in the USA subsequently initiated trials with anti-CD19 CAR T cells in different entities of B cell malignancies such as B cell non-Hodgkin lymphoma (B-NHL) or adult and childhood B cell ALL (B-ALL). In all trials, unprecedented remission rates were achieved and many patients with highly refractory diseases were cured. In 2012, Emily Whitehead, a then 7-year old girl with relapsed, refractory B-ALL was successfully treated by Dr Carl June at the University of Pennsylvania. Her story raised widespread media attention and raised awareness for the potential of CAR T cell therapy. Emily Whitehead is now 14 years old and cancer free for 7 years. With her charity foundation, she supports the development of novel therapies for childhood cancer.
During these initial trials, however, it became evident that the anti-cancer efficacy of CAR T cells comes at the price of drastic systemic toxicities. The most frequently encountered adverse event is the cytokine release syndrome (CRS), a SIRS-like systemic inflammatory syndrome that can be fatal and regularly requires intensive care treatment. Glucocorticosteroids and the IL-6 antagonist tocilizumab can mitigate the toxicity and are now routinely administered in the clinical setting. Additionally, some patients develop a neurological deterioration, termed as CAR-related encephalopathy syndrome (CRES). These symptoms are preliminary in most cases but can be fatal in some. The underlying changes are poorly understood and effective pharmacological treatment options are not yet established.
While the entire pre-clinical and early clinical development of CAR T cell therapy was driven by academic institutions, larger phase III trials required the financial potency and the logistics that only big pharmaceutical companies could provide. Therefore, academic CAR constructs were licensed to Novartis (University of Pennsylvania, tisagenlecleucel, Kymriah®) and Kite Pharma (National Cancer Institute, axicabtagene ciloleucel, Yescarta®). Results of the ELIANA study,26 the first global CAR T cell registration trial in pediatric patients, showed that 41 out 50 patients infused with anti-CD19 CAR T cells achieved complete and durable remissions. Based on these data, tisagenlecleucel received FDA approval for the treatment of pediatric and young adult patients with refractory or relapsed B-ALL (Figure 1).27 Hence, tisagenlecleucel was the first genetically engineered cellular therapy to enter the market. In addition, the JULIET trial28 led to the approval of tisagenlecleucel for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in the USA and Europe.29 Similarly, based on the results of the ZUMA-1 trial,30 axicabtagene ciloleucel gained approval for the treatment of patients with refractory DLBCL, primary mediastinal B-cell lymphoma or transformed follicular lymphoma (Figure 1).31

Challenges and future directions
While clinical experience has validated the unprecedented therapeutic potential of CAR T cell therapy to combat cancer, many challenges persist. For instance, concerns about long-term safety of genetically engineered cells and off-target toxicity remain a topic of debate. The frequency and the severity of adverse events such as infections, CRS and CRES limit CAR T cell therapy to patients that are otherwise refractory to conventional treatment. The main problem with regards to toxicity is the lack of control that treating physicians have over CAR T cells, once they are re-infused into the patients. Different methods to terminate the activity of CAR T cells in case of toxicity or after successful treatment are therefore being developed. A simple approach would be the targeted depletion of CAR T cells with monoclonal or polyclonal antibodies. More elegant approaches use “switchable” CAR T cells that require an adapter molecule to recognize their targets. By modifying the extrinsic administration of the adapter-molecule, physicians would be able to fine-tune the CAR T cell response and adapt to the individual toxicity profile.32
Another main obstacle for the widespread clinical implementation of CAR T cells, not only in developing countries, is the high financial burden that this therapy inflicts on the health care system. With a price range of 71,000−75,000 for a single infusion, novel pricing and reimbursement strategies need to be implemented. Additionally, different approaches exist to cut down the price tag of CAR T cell manufacturing. For example, point-of-care production within academic centers or the development of allogeneic off-the-shelve CAR T cells that do not require the sophisticated logistics of individualized products are promising concepts.
To date, clinical efficacy of CAR T cell therapy is largely limited to hematological malignancies of the B and plasma cell lineage. Other hematological cancers such as myelodysplastic syndrome or acute myeloid leukemia (AML) are more difficult to target due to toxicity against healthy hematopoietic stem and progenitor cells (HSPC). Even more difficult is the translation of CAR T cell therapy for the treatment of solid cancers. In addition to the identification of targetable tumor antigens, impaired access of CAR T cells into the tumor as well as the immunosuppressive tumor microenvironment are major challenges that still need to be overcome.33
In summary, despite certain drawbacks, the CAR T cell story is a paradigm for the successful translation of advancements in basic science for the benefit of critically ill patients. In turn, the unprecedented efficacy of this therapy in the clinical setting has had a fundamental impact on the cancer research community. With lessons learned under “real-world” conditions, scientists around the globe are now working on improving CAR T cell constructs by lowering toxicity profiles, improving control options or reducing manufacturing costs. With the ever-increasing pace of scientific discoveries, the future of immunotherapy in general and adoptive CAR T cell therapy in particular remains to be exciting.