INTRODUCTION
Mycosis fungoides and Sézary syndrome are rare malignancies that belong to the cutaneous lymphomas and are classified within this group as primary cutaneous T-cell lymphomas.1,2 The incidence of cutaneous lymphomas in Europe and the US is estimated to be less than one new case per 100,000 population.3 Mycosis fungoides is considered the most common entity in this group, accounting for about 50% of all cutaneous lymphomas, whereas Sézary syndrome is a rare form, accounting for only approximately 2 to 3%.3,4 Mycosis fungoides most commonly occurs between the ages of 40 and 70 and affects more men than women.5,6 The incidence of mycosis fungoides and its variants is estimated to be between 1:350,000 and 1:110,000 cases, and the mycosis fungoides accounts for 80 to 90% of these cases.6 Sézary syndrome occurs most often in the 5th decade of life and also affects more males than females, and more African Americans than Caucasians.7,8 The annual incidence of Sézary syndrome is less than 1:1,000,000 cases.7,9
CLASSIFICATION
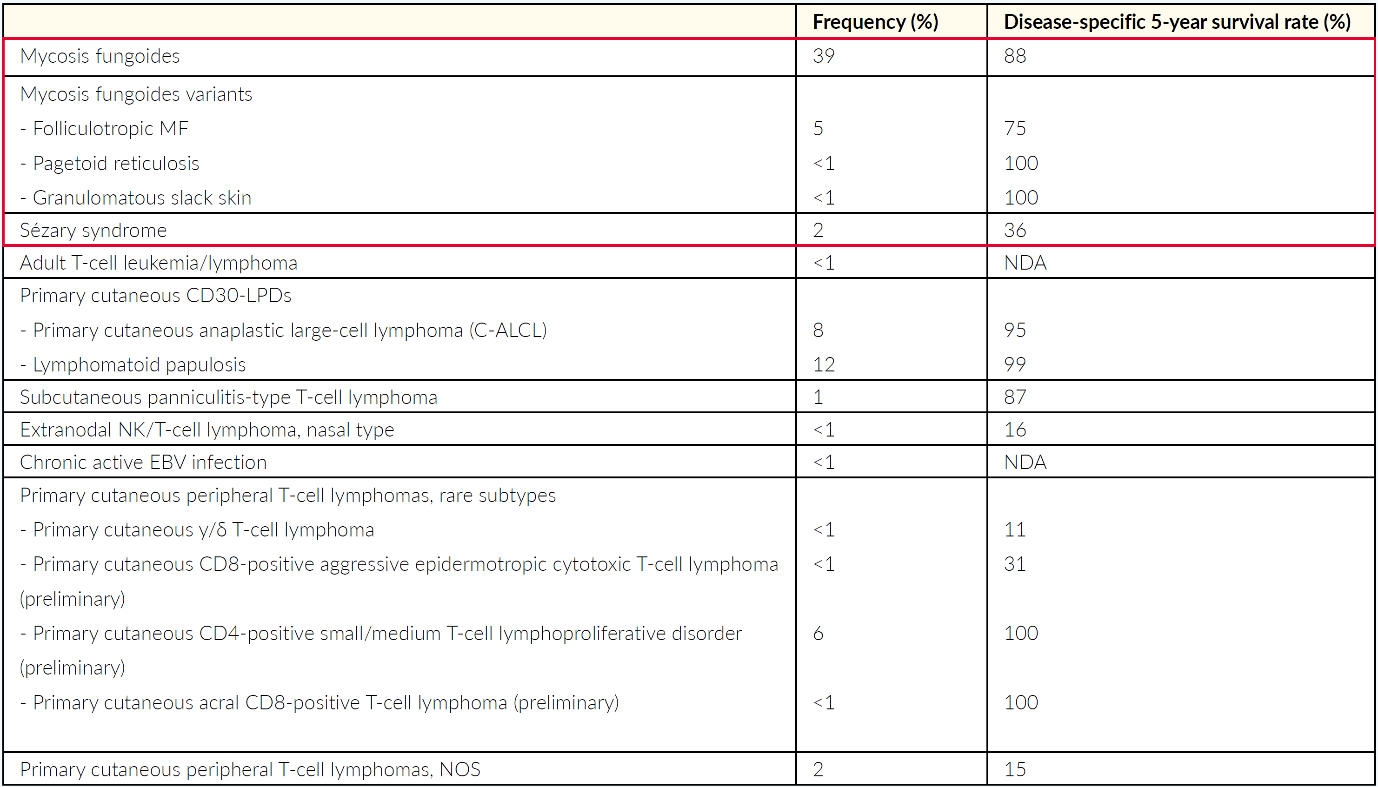
The World Health Organization (WHO) European Organization for Research and Treatment of Cancer (EORTC) classification of cutaneous lymphomas is based on clinicopathological as well as immunohistological and molecular biology criteria.1,2 It therefore also provides a rough estimation with regard to the prognosis of the respective subtype.5 The current version of the WHO-EORTC classification, published in 2018, is shown in Table 1 with an overview of the cutaneous T-cell lymphoma subtypes, including mycosis fungoides and Sézary syndrome.10
.png)
ETIOLOGY/PATHOGENESIS
The etiology of both mycosis fungoides and Sézary syndrome remains poorly understood.6,7 Viruses, such as human T-cell leukemia virus type 1, have been previously suggested as drivers of the disease,11 but recent studies do not present enough evidence to support the viral hypothesis in the pathogenesis of MF and SS.12 Antigen-driven T-cell lymphoproliferation or dyscrasia following medication use,13 as well as genetic factors, i.e., HLA class II alleles predisposing to the disease14,15 have been reported. Recent attempts to profile the genomic landscape of CTCL have demonstrated its high heterogeneity. Although the pathogenesis of the disease cannot be attributed to a small subset of well-defined somatic mutations, copy number variations, fusion proteins, and somatic mutations in diverse cellular and signaling pathways might contribute to the pathogenesis of the disease.15–18 Those include alterations in factors functioning in epigenetic regulation, DNA damage response, cell cycle control, programmed cell death, and T-cell receptor (TCR) signaling, as well as nuclear factor (NF)-κB and Janus kinase (Jak)/signal transducer and activator of transcription (STAT) pathways.17–21 Next to those intrinsic drivers, also extrinsic drivers, most commonly Staphylococcus aureus (SA) and its toxins, are under debate.22 However, epidemiological studies could so far not reliably identify environmental exposure as a trigger for the disease.23
Mycosis fungoides tumor cells arise from so-called “skin-resident effector memory T cells.”24 These cells are located in the peripheral tissues, e.g., in the skin, where they mediate the local immune response independently of the rest of the immune system in the blood and lymph nodes directly on-site as part of the local immune defense.24,25 These cells also survive for a very long time and appear to maintain a stable adhesion to their target tissue. This also explains why the circumscribed skin lesions characteristic of mycosis fungoides often remain restricted to the skin for many years.24,25 In contrast, the malignant cells of Sézary syndrome arise from so-called “central memory T cells,” which typically circulate between peripheral organs, blood and the lymphatic system and thereby play an important role in communication between these compartments.24,25 Accordingly, the tumor cells of Sézary’s syndrome infiltrate the skin diffusely and can also rapidly migrate into the blood and lymph nodes.24,25 Hence, the current understanding is that mycosis fungoides and Sézary syndrome are two distinct entities within cutaneous T-cell lymphomas, differing not only in prognosis but also in pathogenesis.26
CLINICAL PROFILE
MYCOSIS FUNGOIDES
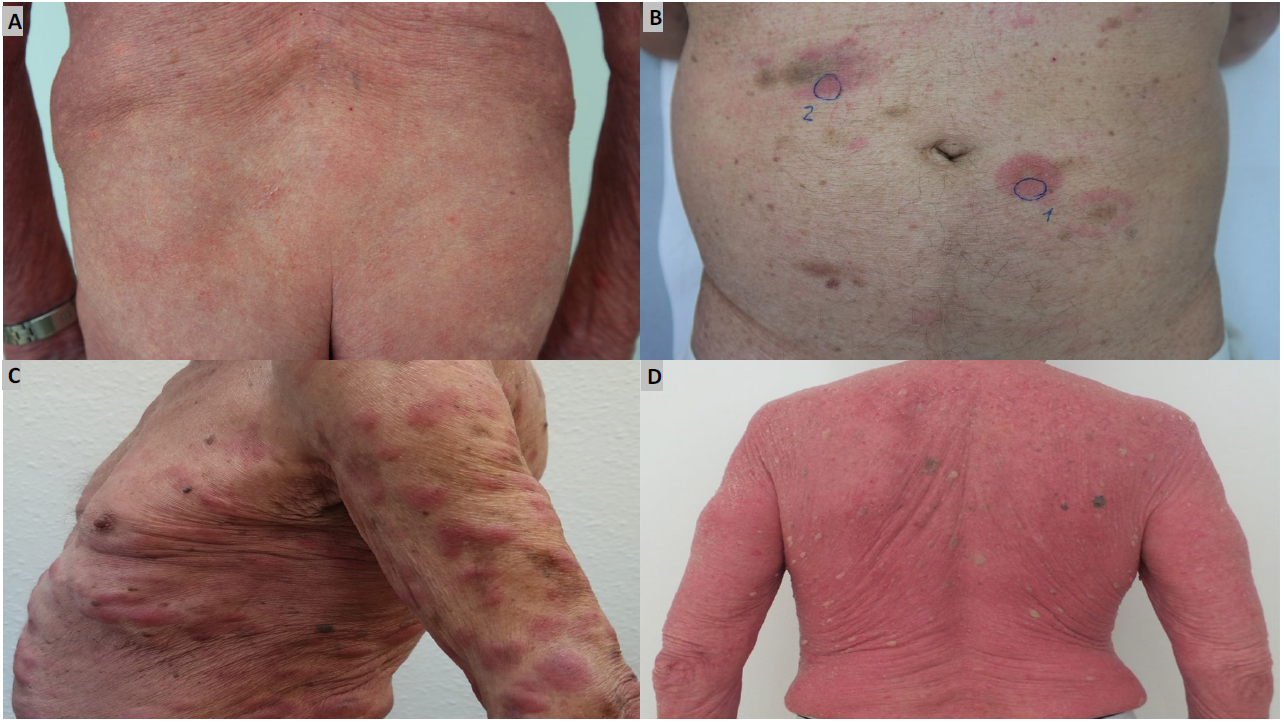
Different clinical stages are distinguished in mycosis fungoides, although not all patients go through each stage. Mycosis fungoides initially manifests as well-circumscribed, erythematous, macular lesions. Sometimes these lesions may appear slightly brownish in color (initial or patch stage), which can remain unchanged for years or decades (Figure 1A).5,26 Since these lesions can mimic other inflammatory dermatoses, the diagnosis of mycosis fungoides is delayed by a median period of up to 36 months after the onset of the initial symptoms.27 The subsequent progression of the disease may vary and manifest as sharply circumscribed, squamous lesions with mild scaling (plaque stage, Figure 1B), tumors (tumor stage, Figure 1C) or erythroderma (Figure 1D).5,26 It is important to distinguish between the latter and Sézary syndrome, since the treatment and prognosis of these two conditions differ.5,26 Folliculotropic mycosis fungoides, pagetoid reticulosis and granulomatous slack skin are distinct subtypes of mycosis fungoides that are differentiated by their clinical and histopathological characteristics clinical progression and prognosis. While the latter two entities are very rare, folliculotropic mycosis accounts for about 5–15% of all mycosis fungoides. Clinically, this subtype has the characteristic grouped follicular papules, preferably in the head and neck region, as well as acneiform lesions and associated alopecia.5
__plaque_stage_(b)__tumor_stage_(c)__e.png)
SÉZARY SYNDROME
Sézary Syndrome is clinically characterized by an erythroderma affecting more than 80% of the body surface.28 Only approximately 25% of patients initially present with the erythroderma, but over 86% develop the characteristic erythroderma during the course of the disease.28 Consequently, patients often present clinically non-specific at the beginning, which leads to a delayed diagnosis of the disease by a mean period of 4.2 years.28 Other symptoms of Sézary syndrome include alopecia, onychodystrophy, palmoplantar hyperkeratosis and intensive pruritus, which is associated with significant distress for the patient.28 The reduction of cutaneous integrity leads to an increased risk of infection by the resident cutaneous flora, e.g., Staphylococcus aureus.29–31 Tumorous skin infiltrates associated with edema as well as hypoalbuminemia may lead to fluid loss. The obligatory findings are generalized enlarged lymph nodes, whereas in the early stages of the disease, often non-specific lymphadenopathy can be detected, in the majority of cases, a specific involvement can be confirmed at a later stage.32
LABORATORY/HISTOLOGY
MYCOSIS FUNGOIDES
In the blood count of patients with mycosis fungoides a significant T-lymphocytosis (mainly CD4+ cells) with increased immaturity of lymphocytes, eosinophilia and atypical T-lymphocytes (Sézary or Lutzner cells) can only be detected in advanced stage IV.6 Occasionally, elevated IgE levels may also be observed.6
Histological examination at the patch stage of the disease shows a variably dense, band-like or lichenoid, subepidermal infiltrate of small, epidermotropic, neoplastic CD4-positive lymphocytes, a mixture of non-neoplastic CD4- and CD8-positive lymphocytes and dendritic cells, however, only scant atypical cells can be found.33,34 A typical feature is the lining-up phenomenon, a pearl string-like accumulation of lymphocytes at the dermo-epidermal junction zone.33,34 Moderate epidermotropism, i.e., migration of atypical lymphoid and monocytoid cells into the epidermis, can be demonstrated.33,34 The migrated cells often appear enlarged in the epidermis and are surrounded by an optically empty halo (halo cells).33,34 A typical spongiosis is absent, as is a pronounced parakeratosis, i.e., a distinction from eczema.33,34 Pautrier’s microabscesses – intraepidermal accumulations of numerous lymphocytes are detectable but occur rather rarely at this stage.35
In the plaque stage, histological examination reveals a dense, band-like infiltrate in the upper dermis consisting of atypical, small or medium-sized lymphocytic cells.33,34,36 Eosinophils and histiocytes may be present in varying density.33,34 Pronounced epidermotropism with Pautrier microabscesses is characteristic of this phase.33–35 The epidermis is frequently acanthotic with parakeratotic keratinization. Adnexotropism, a special form of epitheliotropism, is frequently observed, in which the hair follicles and sweat glands are particularly affected by the influx of infiltrate cells.33–35
Histopathological examination during the tumor stage reveals a dense, diffuse or nodular dermal infiltrate of atypical, medium-sized lymphocytic cells, often involving the subcutaneous fat tissue.37,38 Eosinophils and histiocytes are present to varying degrees.37,38 A high level of mitotic activity may also be observed.6 In approximately 25% of the cases of mycosis fungoides in the tumor stage transform into diffuse, large-cell lymphoma, which may be either CD30-negative or CD30-positive.39 This significantly impairs the prognosis of the affected patients.39,40
SÉZARY SYNDROME
In addition to the clinical findings of pruritic erythroderma and generalized lymphadenopathy, Sézary syndrome is characterized by neoplastic T-cells with cerebriform nuclei and cytoplasmic glycogen granules (Sézary or Lutzner cells) in the blood, skin and lymph nodes. Based on the consensus recommendations of the International Society of Cutaneous Lymphomas and the WHO-EORTC classification, Sézary syndrome can be defined by erythroderma as an obligatory symptom, plus at least two of the following criteria:8,10,41,42
-
A Sézary cell count of >1000/µl in the peripheral blood
-
Immunophenotypic T-cell abnormalities with an elevated CD4/CD8 ratio ≥10, or an increase of ≥30% in CD4+ and CD7- cells, or an increase of ≥ 40% in CD4+ and CD26- cells
-
The presence of a monoclonal T-cell receptor (TCR) population in the blood
Current EORTC proposal:43
-
B0<250/µL
-
B1≥250/µL and <1000/µ
-
B2≥1000/µL CD4+CD7- or CD4+CD26- cells per µL blood
Histological examination of Sézary syndrome is often non-specific. Only in about 50% of cases, the diagnosis of SS can be made histologically. The histopathological features of skin biopsies in Sézary syndrome and in mycosis fungoid skin disease are very similar and thus can hardly be distinguished from each other.44
Histology reveals a significantly increased degree of epidermotropism and more intraepidermal atypical lymphocytes in SS biopsies compared with erythrodermic inflammatory dermatosis (EID). SS show significantly more dermal cerebriform and blastic lymphocytes than EID. Immunohistochemistry reveals a significant loss of CD7 expression in SS case. The lymphocytic infiltrate in SS skin samples express PD-1, MUM-1 and Ki-67. A multivariate analysis identified CD7 loss, increased numbers of small cerebriform lymphocytes, low numbers of CD8(+) lymphocytes and increased proliferation (Ki-67(+) lymphocytes) as the strongest indicators for the diagnosis of SS.44,45
DIAGNOSIS, STAGING AND PROGNOSIS
DIAGNOSTIC APPROACH TO MYCOSIS FUNGOIDES AND SÉZARY SYNDROME
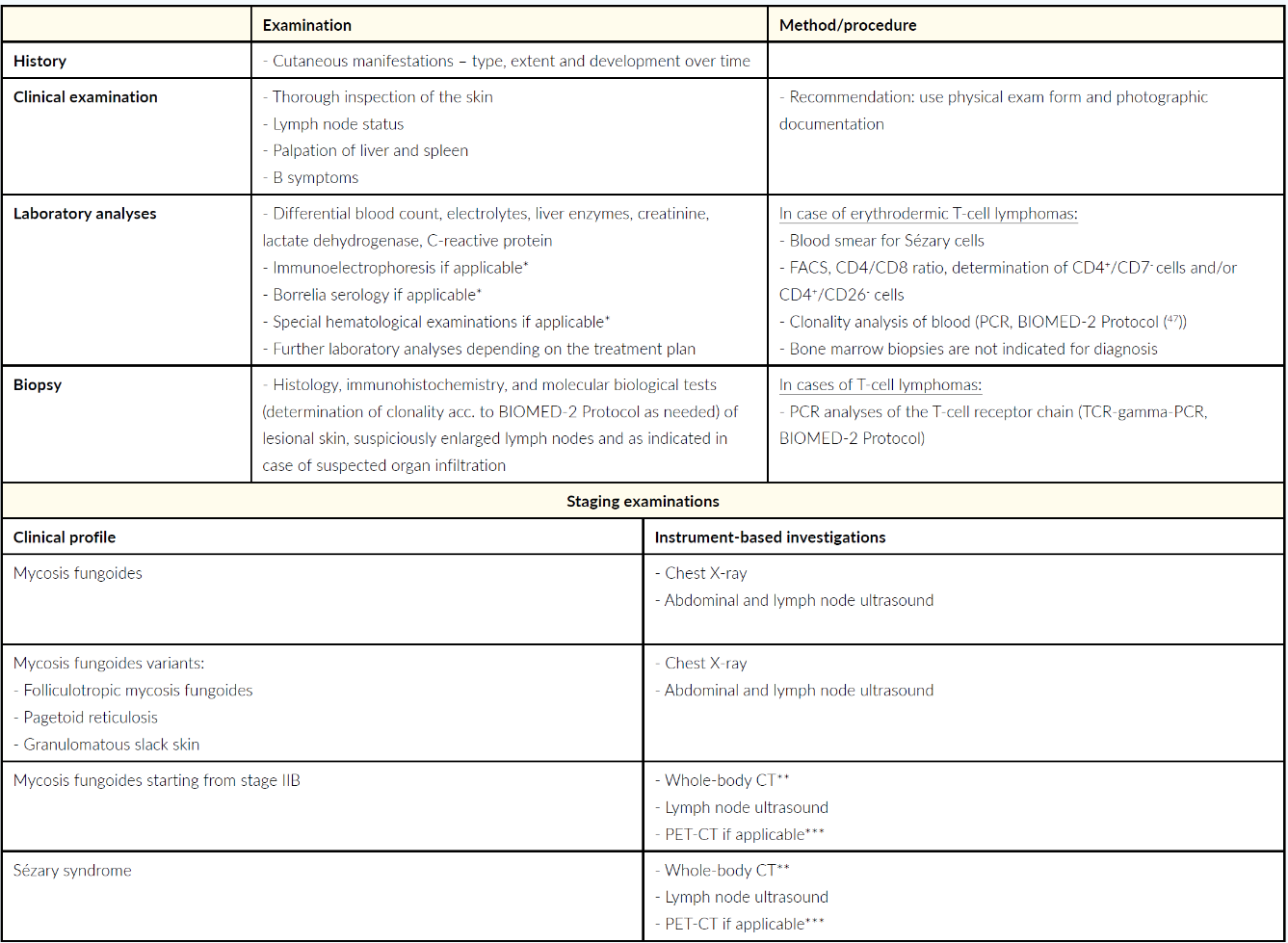
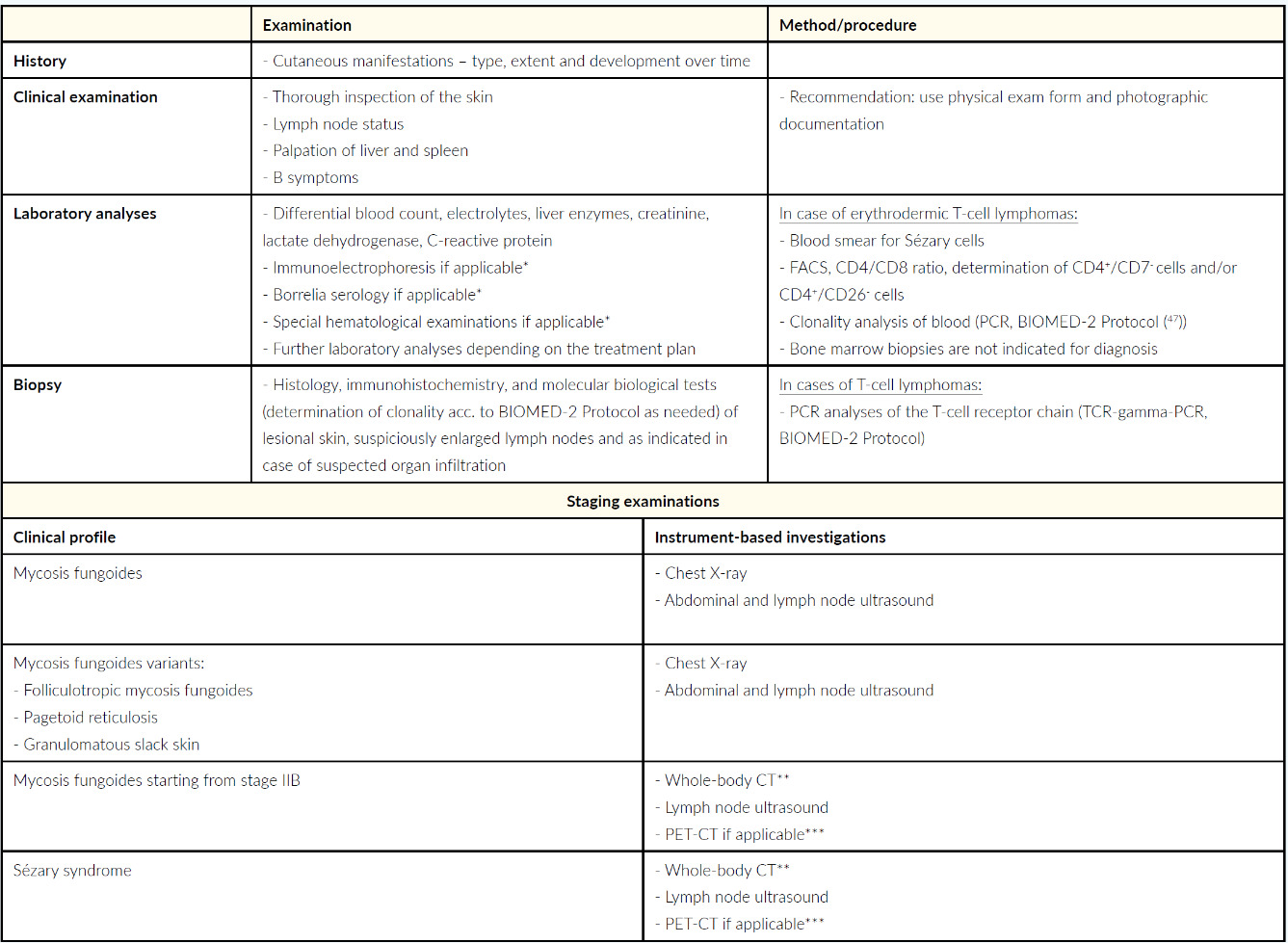
Diagnosis, staging and also the subsequent therapy planned for mycosis fungoides and Sézary syndrome are based on a detailed medical history, precisely documented skin examination, histological examinations (including immunophenotyping and clonality detection if required), laboratory analyses (including flow cytometry or respectively fluorescence-activated cell sorting (FACS). These measures are supplemented by the imaging procedures, such as lymph node ultrasound and whole-body CT.26 The molecular biology and histological findings should not be overestimated compared to the clinical presentation and anamnesis, otherwise there is a risk of overtreatment.26 For example, monoclonality observed during biomolecular testing is also frequently associated with inflammatory disease, the detection of which is therefore often only of limited diagnostic and prognostic relevance.26 The specific recommendations of the Working Group on Dermatological Oncology (Arbeitsgemeinschaft Dermatologische Onkologie – ADO), the German Cancer Society and the German Dermatological Association are presented in the current S2-k Guideline “Cutaneous Lymphomas” covering the diagnostics and staging of mycosis fungoides and Sézary syndrome, depicted in Table 2.1
DIFFERENTIAL DIAGNOSES OF MYCOSIS FUNGOIDES AND SÉZARY SYNDROME
From a differential diagnostic point of view, Sézary syndrome and mycosis fungoides, are difficult to distinguish from each other, since these two conditions share many clinical similarities.8 Examination of peripheral blood involvement is helpful in this regard: findings are absent or minimal in mycosis fungoides, but are an essential component of Sézary syndrome (Sézary cell count in the peripheral blood >1000/µl).8 It is also important to differentiate the initial stage of this disease from parapsoriasis in plaques, microbial or atopic eczemas, psoriasis vulgaris and pityriasis rosea.6 During the plaque stage, mycosis fungoides must be differentiated from cutaneous pseudolymphomas and leukemias, lupus erythematodes tumidus, urticaria pigmentosa and tinea corporis.6 During the tumor stage of the disease, other cutaneous T-cell lymphomas and cutaneous B-cell lymphomas must also be excluded.6 Other, non-neoplastic differential diagnoses of Sézary syndrome include erythrodermic psoriasis, atopic dermatitis or other forms of dermatitis, pityriasis rubra pilaris, adverse drug reactions and idiopathic erythroderma.8 Distinguishing between early-stage Sézary syndrome and erythrodermic inflammatory dermatoses can be challenging.8
TUMOR STAGING AND PROGNOSIS
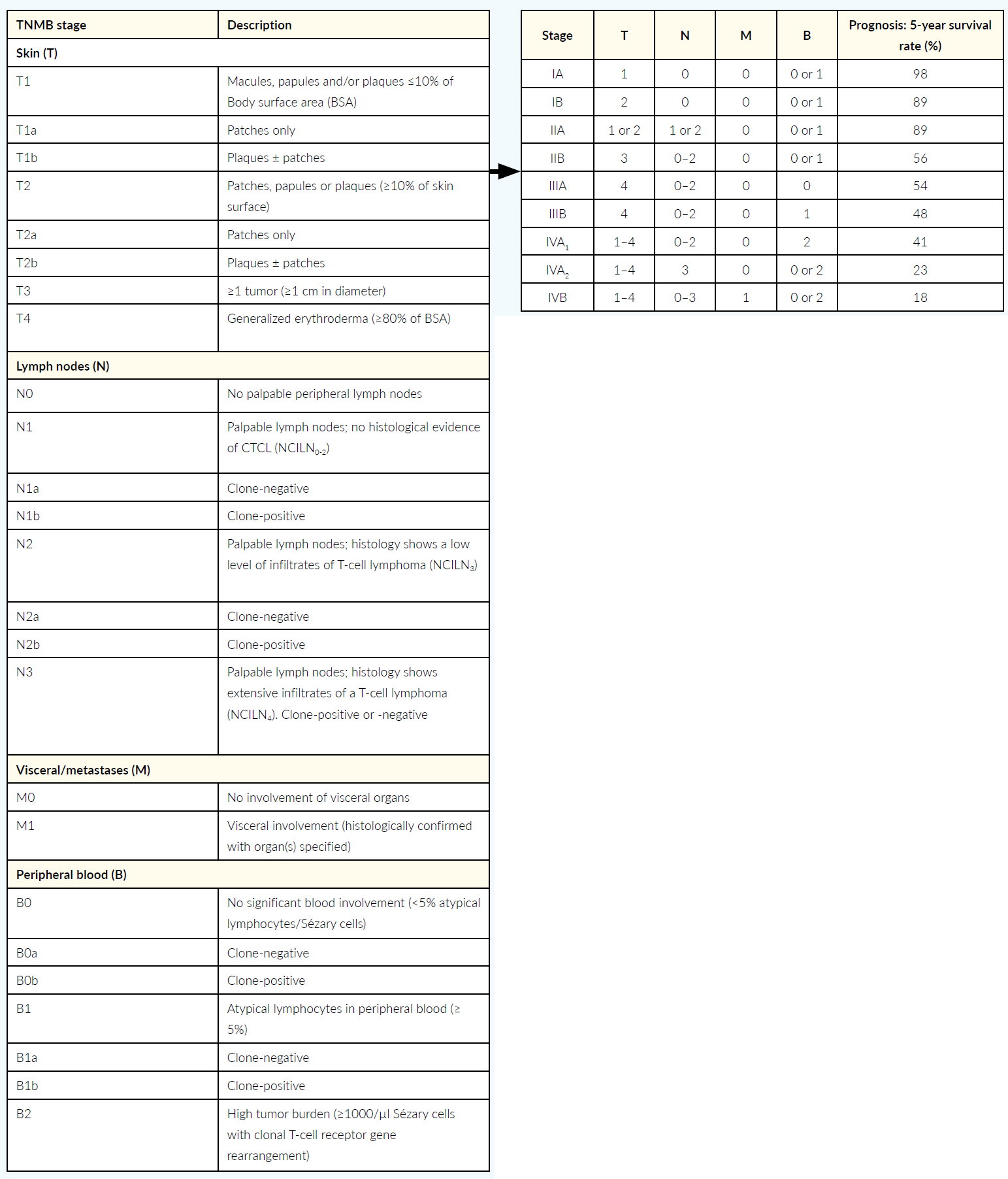
Tumor staging of mycosis fungoides and Sézary syndrome is based on the revised, internationally recognized TNMB classification of the International Society for Cutaneous Lymphomas (ISCL) and the European Organization for Research and Treatment of Cancer (EORTC).46 This classification system includes skin involvement, primary tumor (T), lymph node involvement (N) and distant metastases (M), as well as detection of atypical lymphocytes or Sézary cells in the peripheral blood (B) and is therefore referred as TNMB classification.46 This classification defines the different stages of the disease, and these stages are associated with a certain prognosis, mean 5-year survival rates respectively (Table 3).46,47

In addition to the stage of the disease, there are other factors that also impact the prognosis in patients with mycosis fungoides or Sézary syndrome. A univariate analysis revealed that advanced age, male sex, elevated lactate dehydrogenase (LDH) levels and large cell transformation had a negative impact on survival and increased the risk of disease progression, respectively.48 On the other hand, patients with hypopigmented, poikilodermatous mycosis fungoides or mycosis fungoides with lymphomatoid papulosis have a better chance of survival and a lower risk of disease progression.48 The findings of a multivariate analysis showed, among other factors, that the presence of a tumor clone without Sézary cells in the peripheral blood and folliculotropic mycosis fungoides are independent predictors of decreased survival and an increased risk of progression, while tumor distribution was found to be an independent predictor only for the risk of disease progression.48 In Sézary syndrome, atypical phenotypes and the characteristic loss of the surface markers CD7 and CD26 have also been identified as further negative prognostic factors.8
TREATMENT
LOCAL THERAPEUTIC OPTIONS
The topical drug substances used to treat mycosis fungoides and/or Sézary syndrome include class III-IV topical steroids and local cytostatic such as mechlorethamine. Topically applied bexarotene gel is also applied, a synthetic retinoid analog that presumably interacts with the genes responsible for cell control and thus inhibits tumor cell growth.49,50
Patients with cutaneous lymphoma may also be treated with various forms of phototherapy.1 These therapies include narrow-band UVB treatment (311 nm) or PUVA treatment.1 In the latter form of photochemotherapy, UVA radiation is combined with the administration of psoralens that sensitize the skin to UV radiation and thus enhance the effects.51 Depending on the disease stage and severity, PUVA may be combined with systemic treatments such as oral bexarotene or alpha interferon.1 UV light is also an important component of extracorporeal photopheresis (ECP). ECP starts with taking blood from the patient, from which the leukocytes are isolated by centrifugation (leukapheresis), treated with UV radiation and 8-methoxypsoralen and reinfused.52 The therapeutic effects of extracorporeal photopheresis are based on a modulation of the cellular immune system modulation which leads to a decrease in tumor activity.52 Another therapeutic option for patients suffering from mycosis fungoides is local radiotherapy, typically applied as electron or orthovoltage therapy in a total dose of 4 Gy to 40 Gy for localized, cutaneous B or T-cell lymphomas.1 However, low-dose HDRa brachytherapy can also be used for effective and gentle treatment of sensitive skin areas, e.g., the face, in cases of mycosis fungoides.53 A special form of radiotherapy is total skin electron beam therapy (TSEBT) which is used in conventional (30–36 Gy) and low (10–12 Gy) doses can be applied in mycosis fungoides and Sézary syndrome.1
SYSTEMIC TREATMENT OPTIONS
Systemic treatment options for mycosis fungoides or Sézary syndrome include interferon-alpha, bexarotene, systemic steroids and methotrexate. Alpha interferon, which is used to treat both mycosis fungoides and Sézary syndrome is frequently used in combination with phototherapy, such as PUVA or extracorporeal photopheresis, but it may also be administered as monotherapy to maintain remission.1 A case study of a patient with mycosis fungoides showed that use of alpha interferon after autologous stem cell transplantation maintained sustained complete remission of at least 17 months.54 The retinoid bexarotene, which is used as a topical agent in the earlier stages of the disease, may also be used as a systemic treatment, e.g. in combination with PUVA.1 Systemic steroids or other immunosuppressants such as methotrexate may also be considered in certain cases.3
Antibody-based therapies, e.g., rituximab, alemtuzumab, mogamulizumab or brentuximab vedotin, offer further options for systemic treatment.55 In Switzerland rituximab, alemtuzumab and mogamulizumab can be used as off-label treatments for cutaneous lymphomas. In the EU, mogamulizumab is authorized for the treatment of adult patients with mycosis fungoides or Sézary syndrome, provided that the patients have undergone at least one prior systemic treatment.56 Mogamulizumab, a monoclonal antibody to the type 4 C-C chemokine receptor (CCR4) expressed on tumor cells in cutaneous lymphomas,26 exerts its effects via antibody-dependent, cell-mediated cytotoxicity, i.e. destruction of antibody-bound tumor cells by effector cells of the patient’s own immune system.26 Brentuximab vedotin has approval in the EU and Switzerland for treating patients with CD30-positive cutaneous T-cell lymphoma, provided that the patients exhibited disease progression during systemic treatment or if no other systemic treatment represents a feasible alternative.57,58 Brentuximab vedotin contains a monoclonal, CD30-specific antibody coupled via an amino acid linker to the proapoptotic anti-microtubule agent monomethyl auristatin E (MMAE).59 CD30 itself is a transmembrane cytokine receptor that may be expressed on the surface of tumor cells in different types of non-Hodgkin’s lymphomas, among others.59 In an open-label phase III trial, 131 pre-treated patients with CD30-positive mycosis fungoides or CD30-positive primarily cutaneous anaplastic large-cell lymphoma (pcALCL) were either treated with brentuximab vedotin or a treatment selected by their physician.60 The results showed that a response to treatment with brentuximab vedotin was observed in 56.3% of the patients over a period of at least 4 months, compared to 12.5% in the control group, which is a highly significant difference of 43.8 percentage points (95% CI: 29.1–58.4; p<0.0001).60
In patients with advanced-stage mycosis fungoides or Sézary syndrome, systemic chemotherapies depending on the entity may also be considered. These include mono-chemotherapies with chlorambucil, methotrexate, gemcitabine, liposomal doxorubicin or pralatrexate, or combined chemotherapy regimens, such as EPOCHb or CHOP.1,3,55 Allogeneic stem cell transplantation is the only potentially curative option in patients with advanced-stage mycosis fungoides or Sézary syndrome.5
GUIDELINE-COMPLIANT MANAGEMENT
Treatment of mycosis fungoides and Sézary syndrome depends on the stage of the disease, the prognosis and pre-treatments.1 In general, local, milder forms of treatment are preferentially applied in the early stages with less aggressive course of mycosis fungoides, while, as the disease progresses systemic, more aggressive forms of treatment are increasingly used, and may be combined with local treatment in some cases.1 Mycosis fungoides and Sézary syndrome in Switzerland is mainly treated in accordance with the EORTC guidelines.47
MYCOSIS FUNGOIDES
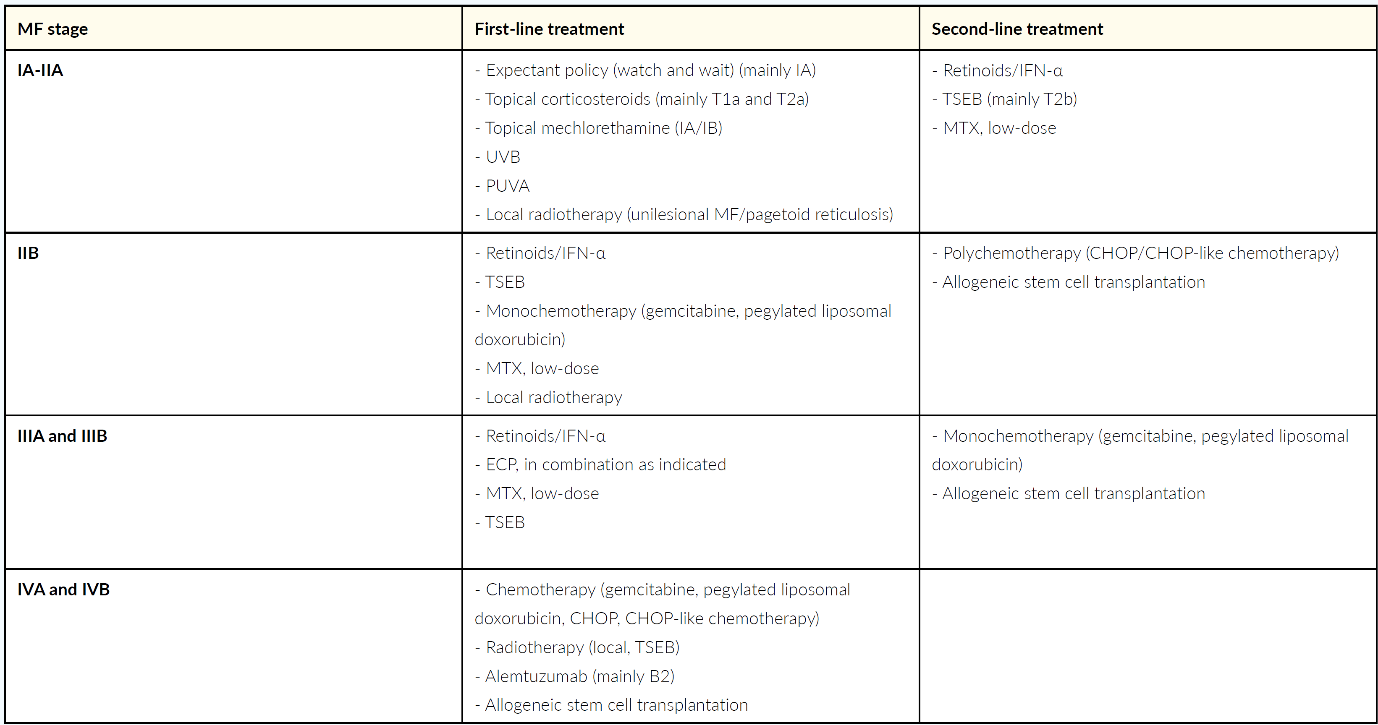
In early stages of mycosis fungoides (IA to IIA) the first/line therapy includes topical steroids, UV treatments (UVB, PUVA), local radiotherapy or topical mechlorethamine.47 As second-line treatments retinoids, such as bexarotene or alpha interferon, low-dose methotrexate, or total skin electron beam therapy are recommended.47 In stage IIB, in addition to retinoids, alpha interferon, total skin electron beam therapy and low-dose methotrexate, which have already been mentioned, potential first-line treatments include monochemotherapy, e.g. with gemcitabine or pegylated liposomal doxorubicin, and localized radiotherapy (add-on therapy).47 In the second line, polychemotherapies (e.g., CHOP) and – in selected patients – allogeneic stem cell transplantation are conceivable options.47 For more advanced disease (IIIA to IVB), extracorporeal photopheresis (IIIA and B), the anti-CD52 antibody alemtuzumab (IVB) and allogeneic stem cell transplantation are further treatment options in addition to the listed therapy options for stage IIB (Table 4).47

SÉZARY SYNDROME
Recommended first-line treatments for Sézary syndrome, which typically follow an aggressive clinical course, are extracorporeal photopheresis (ECP), a combination of chlorambucil and prednisone, systemic treatment with retinoids or alpha interferon combined with PUVA or ECP and low-dose methotrexate.47Potential second-line treatment options include chemotherapy, which is either administered as monotherapy, or as part of a chemotherapy regimen, alemtuzumab and allogeneic stem cell transplantation in selected patients.47
THERAPIES UNDER DEVELOPMENT
The therapeutic landscape for cutaneous lymphomas, such as mycosis fungoides and Sézary syndrome, is changing, with a number of substances currently in various stages of clinical trials. These include, for example, topical resiquimod, which binds to the Toll-like receptors TLR7 and TLR8, inducing an inflammatory response and activating the immune system.59 This immunomodulator has shown promising clinical efficacy and good tolerability in the treatment of cutaneous T-cell lymphomas.59 Several systemic, antibody-based drugs are also currently being tested in clinical trials, e.g., cobomarsen (anti-miR-155 antibody), TTI-621 (anti-CD47 antibody), IPH4102 (anti-KIR3DL2 antibody), atezolizumab (anti-PD-L1 antibody) and alemtuzumab (anti-CD52 antibody), which bind to their target structures and ultimately induce tumor cell death through various pathways.59 In addition to these antibodies, substances with other modes of action are currently being investigated. Duvelisib, an inhibitor of both PI3K-isokinasesc PI3K-δ and PI3K-γ, and E7777, a recombinant cytotoxic interleukin-2 fusion protein that inhibits cellular protein synthesis, as well as talimogene laherparepvec (t-vec), an attenuated type 1 herpes simplex virus that induces an immune response directed against the tumor cell, leading to oncolysis.59 Some of the treatments that are currently under investigation show high response rates, but may also be associated with considerable toxicity.59 Further larger-scale studies are needed to evaluate these forms of treatment.59
FOLLOW-UP
Patients with mycosis fungoides or Sézary syndrome who have stable disease or only exhibit a partial response to treatment, require ongoing treatment. Therefore, the recommendations for follow-up primarily apply to patients in complete remission.1 The primary objective of follow-up in these patients is recognition of recurrence, metastases and secondary lymphomas, as well as any adverse reactions associated to the treatment, e.g. PUVA-related tumors as early as possible so that appropriate measures can be taken if necessary.1 The frequency and form of examinations depend on the stage of the disease.1
CONCLUSIONS
Patients with mycosis fungoides or Sézary syndrome – depending on the entity and stage of the disease – differ considerably in terms of clinical manifestations, prognosis and required treatment.1 Both of these conditions are rare, therefore, patients should preferably be closely monitored in a specialized center by an interdisciplinary team of dermatologists, pathologists, radio-oncologists, oncologists and hematologists.1,3 Such specialized centers can offer diagnostic possibilities in cases that are difficult to classify and can carry out specific, complex therapeutic measures as well as facilitate the patients’ access to respective clinical studies.55 Here, cooperation with the general practitioner is suggested, for instance, the regular laboratory tests required during therapy may also be carried out at a primary care practice.62 Both the specialists from the disciplines involved and the primary care providers thus perform important tasks in the care of patients.
TAKE-HOME MESSAGES
-
Mycosis fungoides and Sézary syndrome, two subgroups of cutaneous T-cell lymphomas, are rare diseases whose etiology is still not fully understood.
-
Mycosis fungoides and Sézary syndrome are considered as separate entities because of the differences in terms of pathogenesis, clinical manifestations and prognosis. Diagnosis, staging and also the subsequent therapy planning for mycosis fungoides and Sézary syndrome are based on a detailed medical history, precisely documented skin examination, histological examinations (including immunophenotyping and clonality detection if required), laboratory analyses (including flow cytometry or respectively FACS). Treatment of mycosis fungoides and Sézary syndrome depends on the disease stage, prognosis and pretreatments. Generally, local forms of treatment are preferred to treat the early, less aggressive stages of mycosis fungoides, while, as disease progresses to advanced stages, and in cases of Sézary syndrome, systemic and more aggressive forms of treatment are used, these may be combined with local treatment in some cases.
a HDR: High Dose Rate
b EPOCH: Etoposide, prednisolone, vincristine (Oncovin®), cyclophosphamide, hydroxydaunomycin
c PI3K: Phosphatidylinositol 3-kinase
COI
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
All authors contributed to and approved the final manuscript.