
INTRODUCTION
Recent technological advances led to the advent of a biomarker-based treatment selection, as well as novel proposed modalities of cancer classification.1–3 The analysis of key genetic features of tumors, as well as that of their surrounding microenvironment and additional individual traits, are increasingly allowing for the tailoring of optimal treatments. Such fine tuning is the core of the increasingly advocated concept of precision medicine.4
The term precision medicine was first introduced in 2011 by the US National Research Council, with the intent of encouraging the definition of molecularly defined subgroups of patients potentially benefiting from targeted therapies.5 The concept was widely embraced since, sustained by public and political efforts such as the Precision Medicine Initiative in 2015,6 and indeed, the number of targeted approaches substantially increased in the past few years. This brought along the need to define a specific and univocal terminology, such as that published by the European Society for Medical Oncology (ESMO) in 2017.7
Here, we discuss some of the most recent examples of precision medicine approaches in oncology, with a focus on the main current and potential histology-agnostic, biomarker-based therapies, as well as the main types of biomarker in use for patients’ stratification. The introduction of novel clinical trial designs is also reviewed.
A PARADIGM SHIFT IN ONCOLOGY: THE INTRODUCTION OF TISSUE-AGNOSTIC INDICATIONS
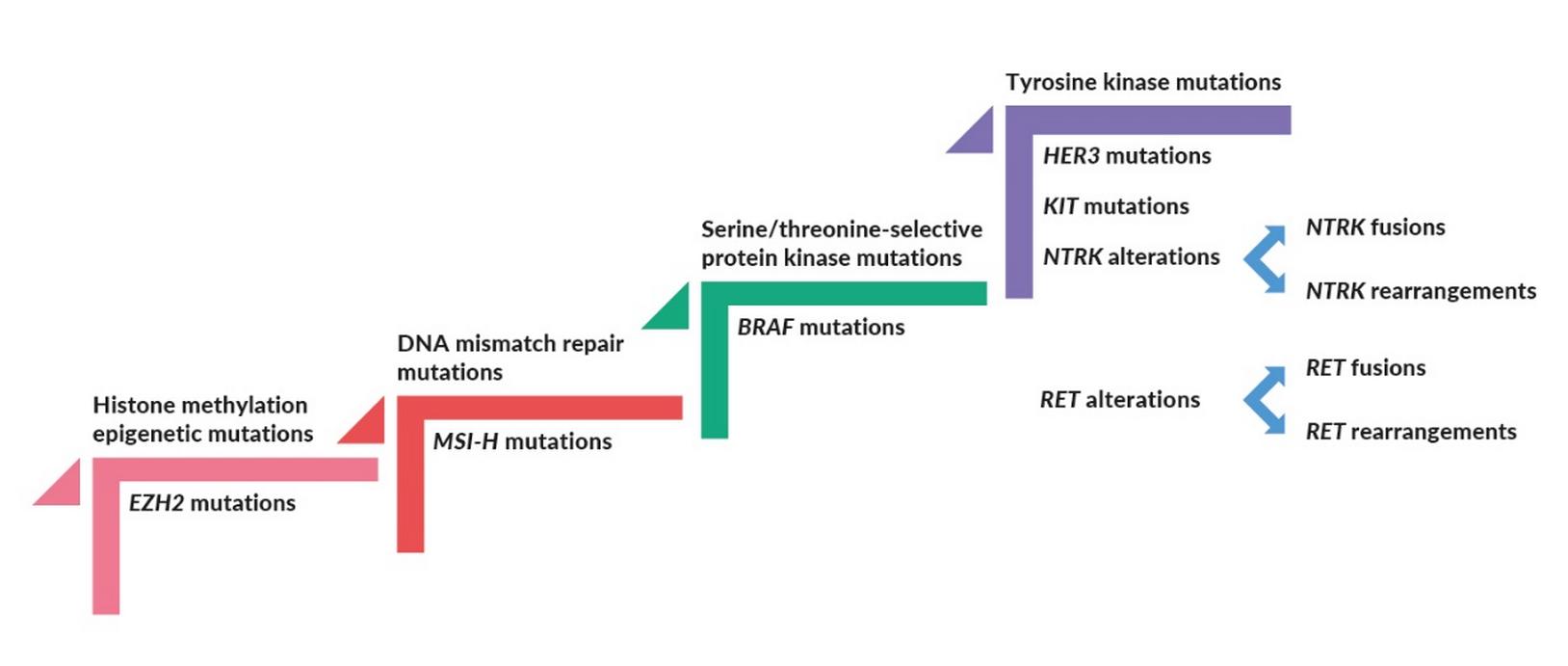
The past few years have witnessed a de facto paradigm shift in oncology, with the U.S. Food and Drug Administration (FDA) approving three drugs for tissue-agnostic, biomarker-based indications, i.e., pembrolizumab, larotrectinib and entrectinib. These drugs leverage on the presence of well-established genomic aberrations.
There are several more gene aberrations that have high exploitable potential (Figure 1). It is plausible to hypothesize that at least a few of those will reach the clinic soon.

The approval of pembrolizumab as a milestone in precision medicine
The approval of the programmed cell death protein 1 (PD-1) inhibitor pembrolizumab for microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) tumors in May 2017 represented the official recognition of the power of biomarker-driven therapies.9
Tumor MSI and dMMR are two predictive biomarkers commonly found together. In fact, the MSI pathway results from a deficient DNA mismatch repair system, generating numerous and repetitive microsatellite DNA sequences.10 In turn, dMMR can be either due to germline mutations, or, more commonly, to epigenetic silencing of the MLH1 (MutL homolog 1) gene. MSI tumors are characterized by an increased mutational and neoantigen load compared to the MSS (microsatellite stable) phenotype, a with concomitant increase in tumor-infiltrating lymphocytes.11 By binding to the PD-1 receptors on T cells, PD-1-targeting therapies disrupt their interaction with their cognate ligands PD-L1 on tumor cells, thereby promoting tumor cell apoptosis.12,13 The novelty of this approach, as opposed to conventional targeted therapies, lies also in the immune nature of the target. The introduction and acknowledgment of pembrolizumab and other immune checkpoint inhibitors brought light to the reviving field of immuno-oncology.14 The Nobel Prize in Physiology or Medicine 2018 awarded to immunologists James Allison and Tasuku Honjo for their discovery of immune checkpoints represents a clear acknowledgment of the importance of this field.
The approval of pembrolizumab in MSI-H/dMMR tumors was based on efficacy data obtained in 5 single-arm multicohort multicenter trials (KEYNOTE-016, -164, -012, -028, and -158), including a total of 90 colorectal cancer patients and 59 patients with other 14 types of cancer.15 The overall response rate (ORR) was 39.6% across all analyzed tumor types (95% CI: 31.7−47.9), with a 7% complete response (CR) rate. The response duration ranged from 1.6 to 22.7 months, with 78% of responses lasting ≥6 months.15
Such tissue-agnostic use of pembrolizumab has not been embraced in Europe or Switzerland to date. Nonetheless, pembrolizumab is approved in Europe and Switzerland in patients with melanoma, non-small cell lung cancer (NSCLC), classic Hodgkin lymphoma (cHL), urothelial carcinoma and renal cell carcinoma (RCC).16,17 In addition, the European Medicines Agency (EMA) approved the use of pembrolizumab in head and neck squamous cell carcinoma (HNSCC) patients.16
Targeting gene fusions towards the goal of precision medicine
Fusions involving neurotrophic-tropomyosin receptor kinase (NTRK) genes are acknowledged drivers of oncogenesis.18 NTRK1, NTRK2, and NTRK3 genes encode the NTRKs TrkA (tropomyosin kinase receptor A or NTRK1), TrkB (NTRK2), and TrkC (NTRK3). In a healthy setting, engagement of the NTRK receptors by their ligand neurotrophin promotes the proliferation and survival of neuronal cells.19–21 The presence of NTRK fusion transcripts results in the constitutive activation of proliferatory signals, thereby driving oncogenesis.18 NTRK fusions were detected in 0.31% and 0.34% of adult and pediatric cancers, respectively, the most common involving NTRK3 (~0.16% of adult tumors) and NTRK1 (~0.14% of pediatric tumors).22 Pediatric melanoma, pediatric glioma and adult thyroid cancers are the ones more commonly expressing NTRK fusions. Despite being present in a limited number of patients, NTRK fusions have been shown to be an optimal therapeutic target. Indeed, agents targeting NRTK fusions showed remarkable efficiency across several cancer types, including distinct soft tissue sarcomas, salivary gland, infantile fibrosarcoma, thyroid, lung, melanoma, colon, gastrointestinal stromal tumor, breast, bone sarcoma, cholangiocarcinoma, carcinoma of unknown primary, congenital mesoblastic nephroma, appendiceal, and pancreas cancers.23
The pan-NRTK inhibitor larotrectinib was investigated in a total of 122 adult and pediatric patients, ranging in age from approximately one month to 80 years, with TRK fusions across 24 unique tumor types. In a first clinical study involving 55 NTRK fusion-positive patients, larotrectinib demonstrated an ORR of 80% (95% CI: 67–90) according to investigator assessment. An astounding 71% of patients maintained tumor regression for more than 1 year.24 An update of this study included new data from the primary dataset, as well as results from a supplementary dataset of 98 patients evaluable for efficacy.23 At up to 26 months of follow-up, the median duration of response in the primary cohort including 44 patients with CR or partial response (PR) was 35.2 months (95% CI: 21.2−not evaluable [NE]).23 The median progression-free survival (PFS) in this dataset was 25.8 months (95% CI: 9.9–NE), with 27 patients showing progression. The combined dataset displayed an ORR of 79% (95% CI: 72–85), with CRs in 16%.23 Importantly, the side effects of larotrectinib appear to be manageable.23 Based on these promising findings, and notwithstanding the fact that studies are still under way, the use of larotrectinib was approved by the FDA in November 2018 for the treatment of NTRK gene fusion-positive tumors.25 Importantly, the EMA gave larotrectinib conditional authorization in October 2019.26 This represented a milestone in the European landscape, as larotrectinib is the first histology-independent cancer treatment approved in the European Union. Of note, to date neither the German Society of Hematology and Medical Oncology (DGHO, Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie) nor Swissmedic endorsed the use of larotrectinib in these tumor-agnostic settings.
A successful path was also followed by entrectinib, a selective tyrosine kinase inhibitor (TKI) of TrkA-C, C-ros oncogene 1 (ROS1) and anaplastic lymphoma kinase (ALK). Similarly to those involving the NTRKs, gene fusions involving ROS1 and ALK are well characterized and clinically actionable across several cancer types.27,28 The presence of ROS1 gene rearrangements was demonstrated most prevalently in NSCLC (1−2% of cases), but also in glioblastoma, ovarian cancer, gastric adenocarcinoma, colorectal cancer and angiosarcoma. ALK rearrangements are also most prevalently found in NSCLC (3−4% of cases),29 but they have been also detected in colorectal, breast, renal cell, renal medullary and thyroid cancers.28 Similarly to the NTRKs, ROS1 and ALK fusions constitutively activate pro-proliferative cellular pathways, thereby promoting carcinogenesis.30,31 Entrectinib has been FDA-approved for ROS1-positive NSCLC in August 2019, and, as of January 2020, is under evaluation by the EMA Committee for Medicinal Products for Human Use.32 To date, entrectinib has not been officially endorsed by the DGHO or the Swissmedic authorities. It should be pointed out that another targeted therapy effective against ROS1-rearranged lung cancers, i.e., the ALK/ROS1/MET inhibitor crizotinib, was previously approved for clinical use in this indication.33 Crizotinib is the only TKI approved by both FDA and EMA for the treatment of ROS1-positive lung cancer. This approval was based on the observation that crizotinib improved ORR and PFS compared to standard of care chemotherapy.34 Of note, crizotinib is also listed amongst the therapeutic options in the DGHO Onkopedia Guidelines.35 For ALK-rearranged lung cancers alone, four targeted therapies have been approved by at least one regulatory agency, i.e., crizotinib, ceritinib, alectinib, and brigatinib.36
The novelty brought by the 2019 FDA approval of entrectinib lies in its additional indication-independent nature, making it the third FDA-approved tissue-agnostic cancer treatment. Entrectinib was indeed authorized for use in adults and pediatric (>12 years of age) patients with NTRK gene fusion-positive solid tumors. This decision, as well as that for ROS1-positive NSCLC, was based on the pooled data from three clinical trials, the phase II STARTRK-2 trial, the phase I STARTRK-1 trial, and the phase I ALKA-372-001 trial,37 as well as data from the phase I/II STARTRK-NG trial.38 The initial data set included 54 adult patients with TRK fusion-positive cancers enrolled in the STARTRK-2, STARTRK-1 and ALKA-372-001 trials.37 The most represented histologies included sarcoma, lung cancer, mammary analogue secretory carcinoma, and breast cancer. The ORR was 57% (95% CI: 43−71), with responses occurring regardless of tumor type and gene fusion (NTRK1 or NTRK3) type. The tumor was completely eradicated in 7.4% of patients. The median duration of response was 10.4 months. As a result, the median PFS and OS were 11.2 and 20.9 months, respectively.37 Seven pediatric TRK fusion-positive cancer patients were subsequently treated with entrectinib in the STARTRK-NG trial.38 The objective response to therapy (>50% target tumor shrinkage) was achieved in all six patients with measurable disease. The responses were durable, with the longest response lasting almost 15 months.38
Overall, these data clearly demonstrate that the use of drugs targeting specific molecules or pathways in selected patient populations, irrespective of the anatomical location of the cancer, constitute a novel powerful avenue to precision medicine in oncology.
BIOMARKERS FOR TARGETED THERAPY SELECTION
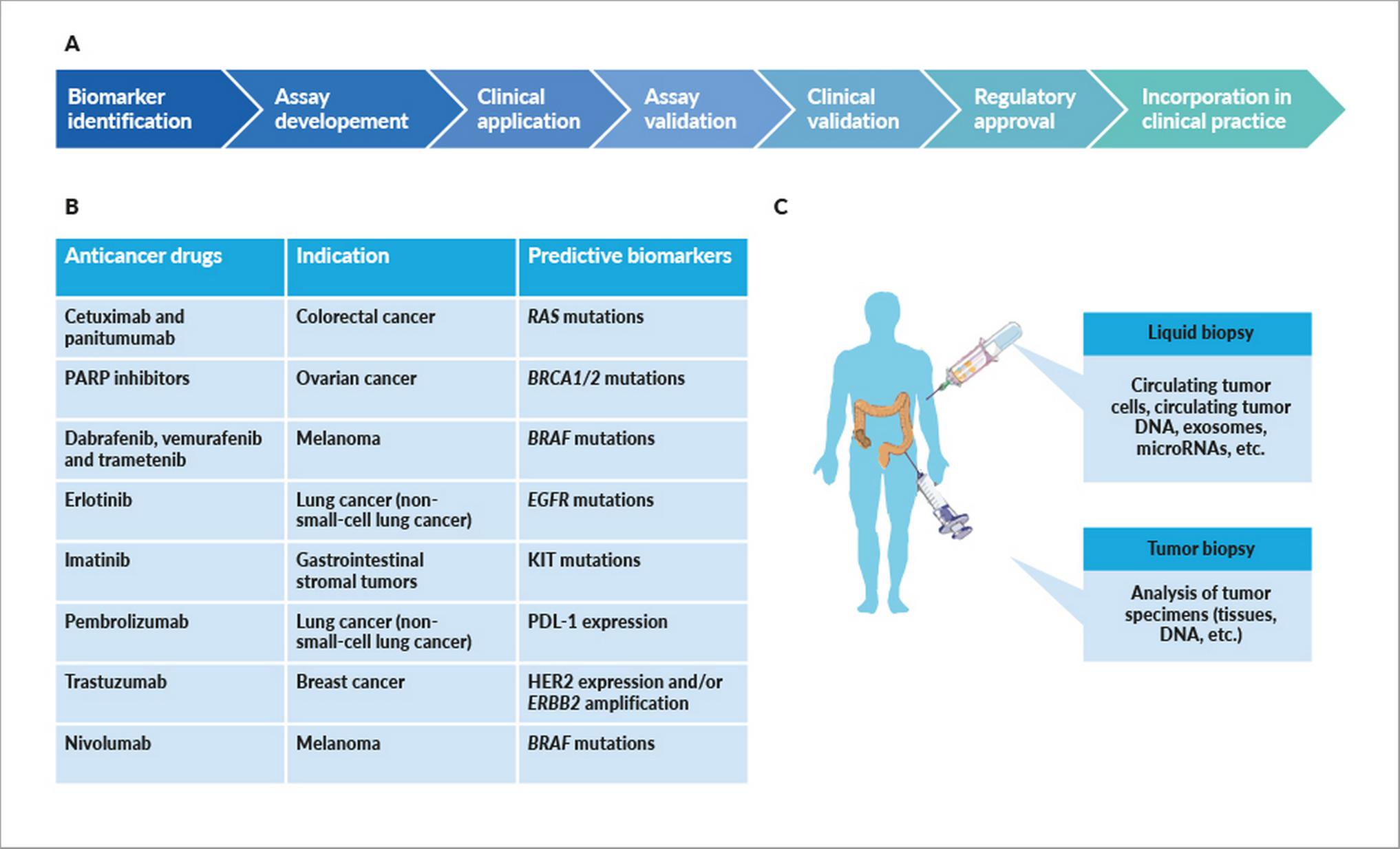
Instrumental in achieving precision medicine is the implementation of biomarkers. A cancer biomarker is a specific feature (DNA, RNA, proteins individually or in grouped in signatures) correlating with either risk of cancer progression (prognostic biomarker) or response to given therapy (predictive biomarker).39 These biomarkers hold the intrinsic ability to identify subgroups of patients, both in terms of survival and therapeutic management. Such patient stratification is as accurate and robust as the underlying biomarker. Thus, the call to precision medicine brings along the need for biomarker development, robust validation and implementation (Figure 2, Table 1).


Typically, cancer biomarkers are divided into biological markers and imaging markers.40,43 Biological markers are assessed in body fluids or tissues, and rely on genomic, transcriptomic or proteomic approaches. The most common liquid specimens are peripheral blood (PB), bone marrow, urine and saliva. PB-derived liquid biopsies have been proposed as novel promising strategies for the early diagnosis of cancer and to define patients’ prognoses.44 Circulating tumor DNA (ctDNA) is fragmented DNA released by tumors and found in the bloodstream and other biological fluids of cancer patients. By possibly recapitulating the entire tumor molecular profile, ctDNA could be used for not only diagnostic, but also prognostic monitoring and therapeutic purposes.44,45 Bone marrow aspirations are often performed to measure the minimal residual disease (MRD) in chronic lymphocytic leukemia.46 Despite offering the advantage of providing more accurate results than peripheral blood measurements, the small withdrawable amounts and the invasiveness of the procedure limits the use of bone marrow draws for MRD measurement.47
Potential biomarkers have been identified in the urine from ovarian cancer patients by metabolomic profiling.48 However, because of their limited performances, urinary markers are not customarily used in clinical practice.49 Salivary biomarkers are also not currently exploited in clinical practice, although evidence exists supporting their potential role in lung cancer diagnosis.50 For solid tumors, formalin-fixed paraffin-embedded (FFPE) tumor specimens are the most used type of sample both for biomarker identification and in vitro diagnostics. Typically, FFPE samples are acquired in routine clinical practice, and represent an accessible source of DNA for further genomic analyses.
Imaging biomarkers are based on radiological techniques such as classic or dynamic contrast-enhanced imaging (MRI [magnetic resonance imaging] and dynamic contrast-enhanced [DCE]-MRI, respectively), mammography, positron-emission tomography (PET), computed tomography (CT), and single-photon emission computed tomography (SPECT). Apart from being able to monitor disease state and progression, computer-guided extrapolation of disease parameters from these tools seems to be the next big revolution in biomarker identification. The term radiomics was first adopted in the past decade to indicate the extraction of high-dimensional quantitative features from medical images, and subsequent mining of correlations between disease diagnosis and/or prognosis.51–53 The increasingly advocated artificial intelligence (AI) approaches are promising sources of novel cancer biomarkers, even more so when combined with underlying genomics, radiomics, pathologic and clinical analyses.54,55
THE ERA OF COMPANION DIAGNOSTICS
By identifying patients that are suitable for treatment, companion diagnostic tests (CDx) are in vitro diagnostic tests supporting the safe and effective use of a specific drug.56 Apart from identifying subpopulations of patients that most benefit from a specific treatment, CDx-based decisions avoid adverse effects in patients unlikely to benefit, simultaneously avoiding the costs associated with adverse events management.
The importance of predictive biomarkers and CDx became evident three decades ago, with the development of trastuzumab for metastatic human epidermal growth factor receptor 2 (HER2) positive breast cancer.57 By binding to HER2, trastuzumab reduces cell duplication rates, and hence their oncogenic potential. The rationale supporting the use of trastuzumab was simple: breast cancer patients who had an amplification of the HER2 gene or an overexpression of the HER2 protein were more likely to benefit from trastuzumab.57,58 Indeed, this proved to be the case.59 An immunohistochemistry (IHC)-based assay detecting HER2 expression was simultaneously developed (HercepTest, Dako), and in September 1998, both trastuzumab and its companion assay were simultaneously approved by the FDA. This was the first regulator-approved combination of drug plus companion diagnostic test to be introduced in the clinic.60 Since then, 37 additional in vitro or imaging CDx have been approved by the FDA for concomitant use with targeted cancer therapies,61 with important positive repercussions on treatment outcome.60 Almost all CDx have been developed for use in classical cancer indications based on the site of tumor origin. So far, the only exception to this is provided by the FDA approval of PD‐L1 IHC 22C3 pharmDx test (Dako, Agilent) concomitantly with that of pembrolizumab. The test proved instrumental to select cancer patients eligible for treatment based on PD‐L1 tumor expression. This represented the first regulatory opening to CDx tests for tissue-agnostic indications, although, thus far, approved cancer indications to go with this test are HNSCC, lung, cervical, urothelial and gastric cancers.62 An in vitro companion diagnostic device for the selection of patients with ROS1 and/or NTRK fusion-positive solid tumors has not yet been clinically validated or approved.
In Europe, the EMA published in 2019 the first guideline on the integration of medical devices into medicinal products, which will be implemented from May 2020 under the new European Regulation on Medical Devices (EU 2017/745).63 Despite the fact that, to date, the EMA does not issue recommendations on CDx, the introduction of such measures is a reflection of the inclusion of CDx as a tool in precision medicine, and the concomitant need for regulation.
Clearly, we’re still witnessing the dawn of the CDx era. Nonetheless, it is evident that the development of appropriate strategies must accompany that of precision medicine, as also demonstrated by the tremendous effort put in by all major stakeholders into CDx development.
BIOMARKER-DRIVEN CLINICAL TRIALS
The increasing understanding of biomarkers and the development of targeted therapies brought along the need for innovative clinical trials designed to evaluate several target-agent combinations in more effective and affordable ways. Three of such new biomarker-driven clinical trials in oncology are basket, umbrella and platform trials (collectively also known as master trials), which assign cancer patients to an arm depending on the targeted aberration found in the tumor.64
Basket trials
A basket trial evaluates one targeted therapy in various cancer types simultaneously. It can be a single- or multiple-arm trial, in which one arm forms a basket that allocates small cohorts with a common single molecular alteration and investigates the efficacy of one targeted agent, regardless of tumor type, histologic type or patient characteristic. The ongoing phase II NCI-MATCH trial is an example of a basket trial. It is composed of 24 sub studies that evaluate at least 17 approved and investigational targeted therapies in previously treated patients with solid tumors, myelomas and lymphomas.65 Recently, the study arm EAY131-H showed clinically meaningful activity for the combination of the BRAF inhibitor dabrafenib with the MEK inhibitor trametinib in heavily pretreated patients harboring BRAF V600E mutations.66 Patients with metastatic NSCLC, melanoma and lymphomas, which are approved indications for this treatment regimen, were excluded from the trial. Other sub studies of NCI-MATCH also reported positive results, including the E131-Y arm which assessed the clinical activity of the investigational AKT inhibitor capivasertib in patients with AKT-mutated cancers.67
The anti-PD-1 nivolumab was first approved by the FDA in 2014 for advanced melanoma, and was subsequently approved for several other indications.68 Of note, it was the first immune-oncology treatment to receive FDA approval based on OS in HNSCC.68 In the NCI-MATCH EAY131- Z1D arm, nivolumab showed promising activity in dMMR non-colorectal cancers of various histopathologic types.69
Less encouraging were the results in the other two NCI-MATCH arms: the first assessed palbociclib in patients with heavily pretreated, advanced non-breast solid tumors and CCND1/2/3 amplification;70 the second tested ado-trastuzumab emtansine (T-DM1) in heavily pretreated patients with HER2-amplified tumors. Nonetheless, T-DM1 demonstrated clinical activity in salivary gland tumors, warranting further studies in this tumor type.71
Umbrella trials
Umbrella trials assess multiple targeted therapies in patients with one cancer type but with different genetic alterations. In this case, the disease is the “umbrella,” under which sub studies for each molecular marker are conducted. For instance, the phase II National Lung Matrix Trial (NLMT) is a current large UK-based umbrella trial in advanced NSCLC patients. By using a Bayesian adaptive design, the trial investigates 8 targeted drugs in 22 molecularly defined cohorts (n=315) based on a 28-gene NGS panel test.72 The first results of the study were presented at the 2019 International Association for the Study of Lung Cancer (IASLC) World Conference on Lung Cancer.73 An interim analysis of 19 cohorts identified several agents with encouraging anti-tumor activity in 4 signaling pathways frequently dysregulated in NSCLC. In the RTK signaling cohort, patients with ROS1 gene fusion and MET exon 14 skipping receiving crizotinib achieved durable clinical benefit (DCB) rates of 71% and 80%, respectively, and an ORR of 68%, with a predictive probability of success (PPoS) of >0.99. In this setting, the EGFR kinase inhibitor osimertinib also demonstrated clinical activity in tumors with the EGFR T790M mutation (DBC rate: 94%; ORR: 76%). In the RAS activation category, the MEK1/2 inhibitor selumetinib plus docetaxel were associated with a DCB rate of 55% and a PPoS of 0.89 for the subgroup with a NF1 mutation. Among patients with a KRAS mutation, the CDK4/6 inhibitor palbociclib showed clinically meaningful results, with a median PFS of 5.4 months and a DCB rate of 44% (PPoS >0.99%). Furthermore, in the subset of patients with cell cycle progression (CCP) aberrations receiving palbociclib, the median PFS exceeded the 3-month target in tumors with CCND1 amplification and tumors with CDKN2A loss. This occurred in both squamous cell carcinoma and adenocarcinoma cohorts, with PPoS ranging between 0.69 and 0.98. No positive result was reported in tumors with CDK4 amplification. In addition, the DCB target has not yet been reached in any of the 4 CCP aberration cohorts. The PI3K/PTEN/AKT/mTOR subgroup appears unlikely to achieve predefined targets of DCB and PFS. For example, among patients with PIK3CA mutation or amplification receiving capivasertib, the DCB rates ranged between 9% and 21%. Finally, the study also demonstrated the impact of smoking and histology on response rates. Never or light smokers achieved response rates of 28% and 17%, respectively, as compared with considerably lower response rates among those with smoking histories of 10−30 pack years (7%) and >30 pack years (10%). Similarly, responses were more frequent in patients with non-squamous than with squamous histology (16% vs 1%).
Platform trials
Platform trials are clinical trials with an open master protocol in which multiple treatments can enter or exit the trial over its course, depending on results observed throughout the study.
One of the first master trials was I-SPY 2, a decade-old yet ongoing platform trial that investigates neoadjuvant treatments in patients with locally advanced breast cancer.74 The trial tests both approved and investigational drugs, and uses an adaptive method to randomize new patients preferentially to the arm that has been best performing in their respective tumor subtype. To date, several agents have graduated to the phase III setting, including the HER-2 receptor inhibitor neratinib and the PARP inhibitor veliparib.75,76 In addition, the study demonstrated that adding pembrolizumab to paclitaxel increased the estimated pathologic CR nearly 3-fold in patients with triple negative breast cancer (60% vs 20%) and patients with hormone receptor (HR)-positive, HER2-negative breast cancer (34% vs 13%).77 The KEYNOTE-522 study presented at ASCO 2019 corroborated these results, showing that the platform trial model might hasten the development of new treatments.78 Further, at 2019 San Antonio Breast Cancer Symposium (SABCS), the I-SPY 2 investigators presented data from a large meta-analysis which showed that the residual cancer burden (RCB) after neoadjuvant chemotherapy is an accurate long-term predictor of disease recurrence and survival across all breast cancer subtypes.79
Hybrid trial designs
The increasing complexity of clinical settings results in an overlap of clinical trial types. For instance, the terms basket and umbrella trial were both proposed to describe the NCI-MATCH trial, which encompasses the evaluation of the use of multiple drugs and the screening of multiple disease populations.80 Another example of this hybrid basket plus umbrella approach is provided by the TAPUR study. This study assesses the off-label use of FDA-approved targeted anticancer therapies in patients with advanced solid tumor, multiple myeloma or B-cell non-Hodgkin lymphoma with a potentially actionable genomic variant.81 Each study cohort includes participants with the same tumor type, genomic variant and study drug.82 Recent data from completed sub studies showed promising anti-tumor activity in NSCLC, breast, and metastatic colorectal cancer. In one study arm, NSCLC patients with CDKN2A loss or mutation achieved responses to palbociclib, which is presently approved for advanced breast cancer.83 In another sub study, pembrolizumab, currently not approved for this indication, demonstrated anti-tumor activity in heavily pre-treated patients with advanced breast cancer and high tumor mutational burden.84 In addition, 3 cohorts enrolling patients with colorectal cancer reported positive results,85–87 with more than 30 other sub studies showing evidence of anti-tumor activity.
CONCLUSIONS
The ever-growing discovery of new therapeutic targets, as well as the recognition of the role of the immune system in shaping tumor development, paved the way to novel, promising approaches to contrast and hopefully cure cancer. The introduction and clinical use of targeted therapies notably reduces treatment-related toxicity and, apart from being more effective, they often offer the possibility of oral dosing.
It is widely accepted that the only way to truly achieve clinically encouraging results is via the implementation of precision medicine approaches, i.e., patient stratification. Instrumental to this undertaking is the adoption of reliable biomarkers to guide treatment decision. Before patients can be started on a selected treatment, the presence of a specific mutation in the tumor should be confirmed by a validated test. The effort put in the development, implementation and regulation of CDx is a clear indication of the current need to address this issue. Precision oncology drug development has become possible also thanks to the introduction of faster tracks to the clinic. Tailored clinical trial formats, as well as breakthrough therapy designation and accelerated approval processes have recently become valuable tools supporting this paradigm shift.
Taken together, these observations demonstrate how the latest scientific, technological and methodological advances are taking us increasingly closer to the ideal concept of personalized medicine in oncology.