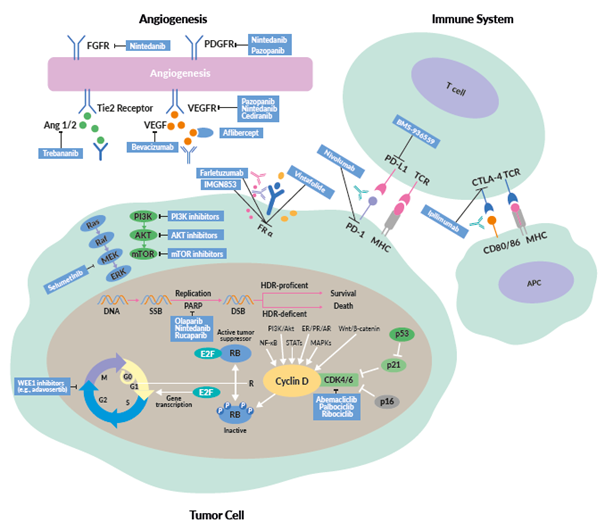


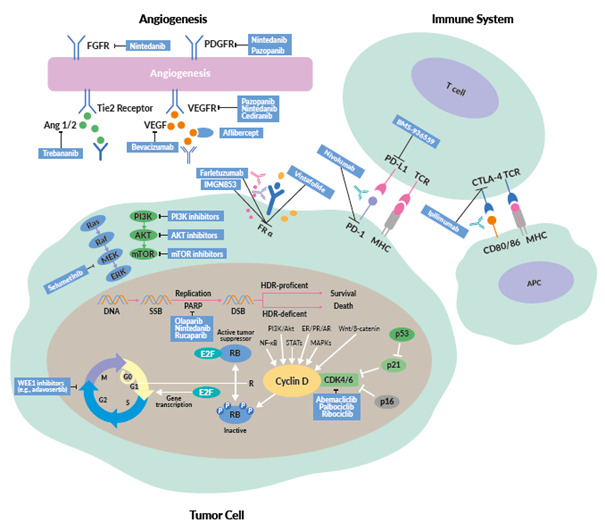
INTRODUCTION
Despite being the third most common gynecologic cancer, ovarian cancer (OC) still has a poor prognosis. However, due to the recent oncologic advances, survival outcomes of patients with OC have been improved in the past years.1 There are several reasons for this development, including better surgical procedures, systemic therapy and modern targeted therapy. More than 70% of OC patients are diagnosed in the advanced International Federation of Gynecology and Obstetrics (FIGO) stage III or IV, which implicates that cancer has spread to the abdominal cavity or the distant organs.2 The median 5-year overall survival (OS) rates in this patients population range between 30 and 40%. Only the minority of OC patients can be cured at relapse. However, recent data (DESKTOP III Study) demonstrated a survival benefit in highly selected patients with relapsed OC if the recurrent disease can be removed and chemotherapy is applied.3
Nowadays, five distinct histologic subtypes are defined, including high-grade serous ovarian cancer (most common subtype), low-grade serous ovarian cancer, mucinous ovarian cancer, clear cell ovarian cancer and endometroid ovarian cancer.4 Treatment includes optimal staging and maximal cytoreductive debulking followed by chemotherapy with 6 cycles of carboplatin/paclitaxel.3 For high-risk homologous repair deficiency (HRD), the addition of olaparib and bevacizumab is the current standard of care, while for BRCA-mutated cancer, single-agent olaparib is the current standard treatment. This is due to the lack of direct comparison of olaparib plus bevacizumab and olaparib as single-agent maintenance in the PAOLA-1 study.5 In the relapsed setting, poly(ADP-ribose) polymerase (PARP) inhibitors (i) are approved as maintenance therapy, regardless of BRCA mutation or HRD status.6 In patients with recurrent disease, chemotherapy is still used according to the Gynecologic Cancer InterGroup (GCIG) in case of platinum-sensitive disease (platinum-resistant: platinum-free interval (PFI) <6 months; partially platinum-sensitive: PFI: 6−12 months; platinum-sensitive: PFI >12 months), although there is a newer GCIG definition of response to platinum-based therapy including the CA-125 kinetics.7 Recent data showed that the use of the PARP inhibitor olaparib in combination with cediranib, a multi-kinase inhibitor, is as effective as platinum-based chemotherapy.8
This review provides an overview of new treatment options in OC, including the latest data on PARP inhibitors, antiangiogenic drugs, immunotherapy and targeted therapies (Figure 1).
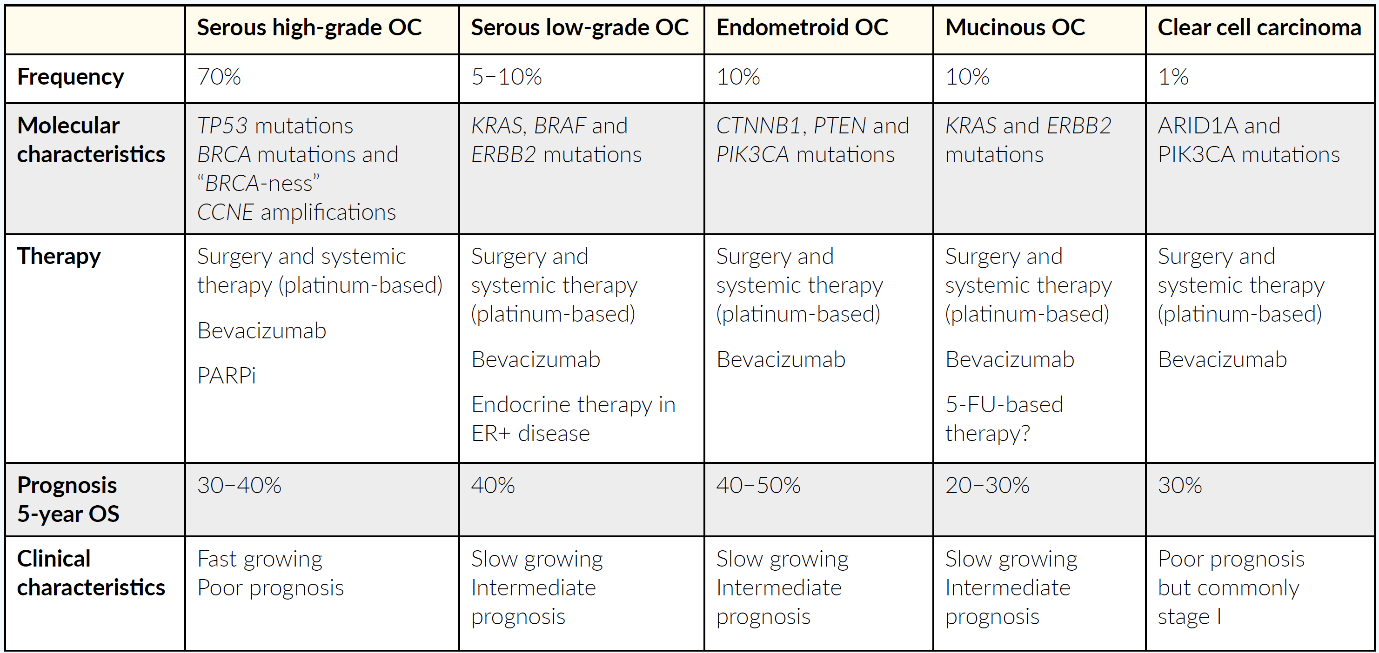
MOLECULAR SUBTYPES OF OC AND TREATMENT INDICATIONS
Currently, all advanced OC subtypes are treated similarly, although the subtypes have different molecular characteristics and clinical outcomes. The most common subtype is serous high-grade carcinoma typically characterized by genomic instability.10 The disease has a poor clinical outcome and is commonly diagnosed in advanced FIGO stage III/IV. Tumors frequently harbor TP53 signatures and BRCA mutations. Around 20% of patients have BRCA mutations, while approximately 50% of patients have a defect in the HRD mechanism. These molecular changes have prognostic and predictive potential. Indeed, HRD and BRCA are established biomarkers for the use of PARPi, which changed the treatment paradigm in high-grade serous ovarian cancer.11
Other subtypes include low-grade serous carcinoma, endometrial, mucinous, clear cell, and mixed histology.4 Table 1 provides an overview of the molecular landscape, treatment and clinical outcomes in all OC.
_subtypes__molecular_characteristics_and_the_current_standard_therapy.png)
CURRENT AND EMERGING TREATMENTS
STANDARD OF CARE: CYTOREDUCTIVE SURGERY AND ANTIANGIOGENIC THERAPY
Despite new systemic treatment options, one of the most important therapy in advanced OC remains complete cytoreductive surgery.3,12 Data showed the better the tumor has been removed, the better the outcome for patients with OC. Optimal debulking is defined by the GCIG consensus as the absence of macroscopic residual disease (R0).7 After optimal surgery, state-of-the-art therapy includes 6 cycles of chemotherapy with carboplatin (area under the curve [AUC], 5−6) and paclitaxel (175 mg/m2 every 3 weeks), followed by an adequate maintenance strategy. If available, bevacizumab, an antiangiogenic drug, is the standard of care in all patients not receiving an optimal cytoreductive surgery, based on data from the phase III ICON7 trial.13 A primary analysis of ICON 7 showed an OS benefit of around 9.5 months with bevacizumab versus standard therapy in around 500 patients with visible residual disease (R1) or FIGO stage IV.
CURRENT TREATMENT LANDSCAPE WITH PARP INHIBITORS
The addition of a PARPi depends on the clinical situation, biomarker and subtype of ovarian cancer. Olaparib was the first approved PARPi in relapsed and advanced serous high-grade OC with a pathological BRCA1 or BRCA2 mutation. In Study 19, 265 patients with relapsed high-grade serous OC after finishing re-induction platinum-based chemotherapy underwent 1:1 randomization to receive either olaparib (n=136) or placebo (n=129).14 In the initial presentation of the intention-to-treat (ITT) population, the median progression-free survival (PFS) was almost doubled with olaparib (8.4 months vs 4.8 months with placebo; HR: 0.35; p<0.00001). A post hoc analysis further demonstrated significantly improved PFS in olaparib-treated patients with BRCA mutations (median, 11.2 vs 4.3 months with placebo; HR: 0.18; p<0.0001) and in BRCA wild-type patients (7.4 vs 5.5 months; HR: 0.54; p=0.0075).15 Similarly, in the confirmatory phase III SOLO2 trial, patients with deleterious BRCA mutations achieved a significantly improved PFS with olaparib.16 In the final analysis of the study, the median OS was 51.7 months with olaparib and 38.8 months with placebo (HR: 0.74 [95% CI: 0.54−1.00]; p=0.0537), at a median follow-up of approximately 65 months.17 The most common grade ≥3 treatment-emergent AE was anemia (olaparib: 21%; placebo: 2%), while serious treatment-emergent AEs were reported in 26% of olaparib-treated and 8% of placebo-treated patients.
Olaparib as the first-line maintenance therapy was assessed in the randomized, phase III SOLO1 trial in patients with newly diagnosed advanced high-grade serous or endometrioid OC with a mutation in the BRCA1/2 genes who had a complete (CR) or partial (PR) clinical response to platinum-based chemotherapy.18 In total, 391 patients underwent 2:1 randomization to receive olaparib tablets (300 mg twice daily) or placebo for up to 2 years or until disease progression. After a median follow-up of 41 months, the median PFS was not reached with olaparib and was 13.8 months with placebo (HR: 0.30 [95% CI: 0.23−0.41]; p<0.001). This PFS benefit was sustained at 5 years, with a median PFS of 56.0 months among olaparib-treated and 13.8 months among placebo-treated patients, resulting in a 5-year PFS rate of 48.3% and 20.5%, respectively.19
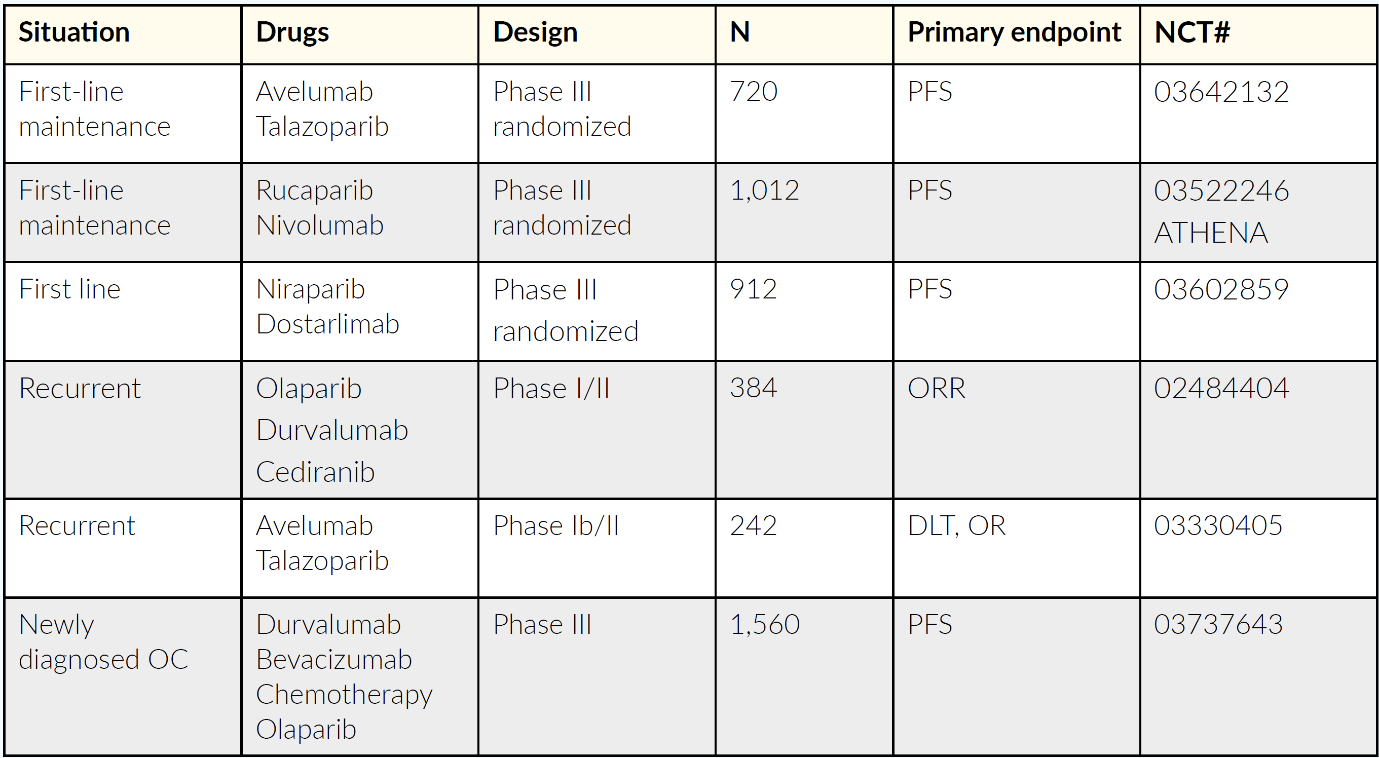
In the last years, several strategies combining the most promising agents, including PARPi, antiangiogenic agents and immunotherapy, have been developed (Table 2). A phase II study that assessed cediranib plus olaparib versus olaparib alone in patients with relapsed platinum-sensitive ovarian cancer showed a significantly longer PFS with the combination therapy (16.5 months vs 8.2 months, HR: 0.50; p=0.007).20 Compared with historical chemotherapy cohorts, cediranib plus olaparib appeared to be very effective, and a randomized phase III trial was set up.8 This study compared olaparib alone and in combination with cediranib, a potent inhibitor of VEGF receptor tyrosine kinases, with platinum-based chemotherapy in patients with recurrent platinum-sensitive OC. The median PFS was 10.3 months with chemotherapy, 8.2 months with olaparib and 10.4 months with olaparib plus cediranib, with response rates of 71.3%, 52.4% and 69.4%, respectively. The primary endpoint of the study was not met as the combination of olaparib and cediranib could not be shown to be superior.8 A further investigation of chemotherapy followed by the combination of olaparib plus cediranib combination is needed. Indeed, an ongoing ICON9 study aimed to assess the efficacy of maintenance treatment with olaparib plus cediranib versus olaparib alone following response to platinum-based chemotherapy in women with platinum-sensitive ovarian, fallopian tube or peritoneal cancer during the first relapse.21

The combination of olaparib and bevacizumab is the standard first-line maintenance therapy for patients with HRD-positive OC, based on data from the PAOLA trial.22 In this study, 806 patients with advanced high-grade serous ovarian cancer were treated with bevacizumab maintenance according to the current standard and were randomized 2:1 to receive either olaparib (n=537) or placebo (n=269). After a median follow-up of 22.9 months, the median PFS was 22.1 months with olaparib plus bevacizumab and 16.6 months with placebo plus bevacizumab (HR: 0.59 [95% CI: 0.49−0.72]; p<0.001). Patients with tumors positive for HRD, including BRCA mutations, achieved the greatest PFS benefit with olaparib (median PFS, 37.2 months vs 17.7 months with placebo; HR 0.33), while patients with HRD-positive tumors without BRCA mutations had a median PFS of 28.1 months with olaparib and 16.6 months with placebo (HR: 0.43).
For patients with no access to bevacizumab or with a contraindication to antiangiogenic agents, the first-line niraparib might be an adequate treatment option.23 In the randomized, phase III PRIMA study, niraparib showed a significant benefit for patients with newly diagnosed advanced OC with a high risk for relapse, as defined by residual cancer burden.24 Overall, 733 patients were randomized 2:1 to receive either niraparib or placebo once daily, following a response to platinum-based chemotherapy. The primary endpoint was PFS in patients who had tumors with HRD and in the overall population. The median PFS was significantly longer in patients with HRD-positive tumors receiving niraparib than in those receiving placebo (21.9 months vs 10.4 months; HR: 0.43; p<0.001). In the overall population, the median PFS was 13.8 months and 8.2 months (HR: 0.62; p<0.001). This PFS benefit was consistent in all key subgroups, including BRCA-positive and BRCA-negative/HRD-negative. The most common side effects of niraparib were anemia, neutropenia and thrombocytopenia.
Niraparib was also assessed in 553 patients (gBRCA: n=203; non-gBRCA: n=350) with platinum-sensitive, recurrent OC in the randomized, double-blind, phase III ENGOT-OV16/NOVA trial.25 At data cutoff, niraparib was associated with significantly longer median PFS (gBRCA: 21.0 months vs 5.5 months with placebo; HR: 0.27; non-gBRCA: 9.3 months vs 3.9 months; HR: 0.45). In non-gBRCA patients who had tumors with HRD, the median PFS was 12.9 months with niraparib and 3.8 months with placebo (HR: 0.38). In the final analysis, which assessed the safety and long-term efficacy of niraparib, all patients showed benefit on treatment with niraparib beyond the first progression (gBRCA: HR: 0.67; non-gBRCA; HR: 0.81).26 In the gBRCA cohort, the median OS was 43.6 months with niraparib versus 41.6 months with placebo (HR: 0.93). After the inverse probability of censoring weighting adjustment (IPCW), the median OS was 43.8 months with niraparib compared with 34.1 months with placebo (HR: 0.66). At an average follow-up of 5.6 years, the median OS was also improved in the niraparib versus the placebo arm in the gBRCA cohort (43.6 months vs 41.6 months; HR: 0.93). After the IPCW, the median OS was 43.8 months with niraparib compared with 34.1 months with placebo (HR: 0.66).
PARPi are considerably more effective in tumor cells defective in homologous recombination-based repair, as compared with HR competent counterparts, which represents an area of unmet medical need. In the subgroup of patients with homologous-recombination proficiency in the PRIMA study, the PFS benefit was only modest with niraparib versus placebo.24 As a result, niraparib in the first-line maintenance setting has been approved in Switzerland only for the treatment of patients with HRD-positive tumors.27
IMMUNOTHERAPY
Several different immunotherapy approaches have been undertaken to improve clinical outcomes in patients with epithelial OC, by enhancing the immune response; however, none of these methods have been already approved by medical drug authorities. For example, various types of vaccination strategies have been tested.28 These include anti-idiotypic antibody-based vaccination (e.g., abagovomab); peptide- and protein-based vaccination (e.g., NY-ESO peptides); lymphocytes-based vaccination (e.g., autologous LAK plus IL2); carbohydrate-based vaccination (e.g., MUC1-Sialyl-TN); DNA plasmid-based vaccination (e.g., poxviral vector PANVAC-V); combination-based vaccination (e.g., with sunitinib); vaccination based on dendritic cells (e.g., autologous DC pulsed with MUC1-derived peptides or HER-2/neu). However, none of these approaches are currently in clinical practice because they failed further clinical development.
Tumors with a high mutational load, such as melanoma, kidney or lung cancer, are ideal candidates for therapies using immune checkpoint inhibitors (ICIs), which led to approvals of several ICIs in these cancers.29,30 So far, the role of single-agent immune checkpoint inhibitors in OC is not fully clear, although the PD1/PD-L1 pathway seems to play an important role. In OC, PD-1 is expressed on TILs.31 A study showed that higher versus lower tumor PD-L1 expression is associated with a significantly poorer prognosis.32 In a preclinical model, inhibition of PD1 and PD-L1 demonstrated tumor rejection and reprogramming of the tumor microenvironment.33 In one of the first phase I study assessing an anti-PD-L1 antibody in patients with selected advanced cancers, objective responses were also observed in OC.34 Data from phase II studies, which investigated single-agent immune checkpoint inhibitors (ICIs) in OC patients with the platinum-resistant disease and poor prognosis, demonstrated low response rates but higher disease control rates (DCR) and long-term remissions (Table 3).34–37

The assessment of PD-L1 as a prognostic marker in OC is less clear. Data from Hamanishi et al. (2007) showed that PD-1 was not an optimal prognostic marker.32 More importantly, the mutational landscape might be important to select the right treatment for the suitable tumor. In general, the mutational burden is lower in OC than in other cancers. However, ovarian tumors with germline or somatic BRCA1/2 mutations were found to have a higher frequency of exome mutations (median, 67.5) than tumors with wild-type BRCA (median, 49.5).38
Further development includes the combination of an ICI with chemotherapy, PARPi or bevacizumab. Recently, the primary results of the IMagyn050/GOG 3015/ENGOT-OV39 study, a double-blind, placebo-controlled randomized phase III trial of bevacizumab-containing therapy with or without atezolizumab in newly diagnosed stage III/IV OC, were presented.39,40 All patients (n=1,301) received carboplatin (AUC6) and paclitaxel (175 mg/m2) every 3 weeks for 6 cycles plus bevacizumab (15 mg/kg; every 3 weeks in cycles 2−22). The patients were randomized 1:1 to receive either atezolizumab (1,200 mg every 3 weeks) or placebo. The primary endpoint was not met as atezolizumab was not associated with significantly improved PFS in the ITT population (median, 19.5 months vs 18.4 months with placebo; HR: 0.92; p=0.27). In the subgroup of patients with PD-L1 immune cell (IC) ≥5%, there was a statistically significant difference in PFS between the atezolizumab and placebo arms (20.2 months vs not estimable [NE]; HR: 0.64; p=0.0278).
For patients with OC in the recurrent setting, the combination of a PARPi and ICI seems to be promising. Recent studies have demonstrated a synergistic effect of PARPi and ICIs; PARPi can stimulate interferon genes (STING)-dependent manner and is augmented by immune checkpoint blockade.41 Recently, the phase I/II TOPACIO/KEYNOTE-162 assessed the combination of the PARPi, niraparib, and the ICI, pembrolizumab in patients with triple-negative breast cancer or recurrent OC, irrespective of BRCA status.42 Overall, 62 patients received 200 mg daily niraparib plus 200 mg intravenous pembrolizumab on day 1 of each 21-day cycle. In the pooled OC cohort, ORR was 18%, including a CR rate of 5% (n=3), with a DCR of 65%. The median duration of response (DoR) was not reached (range, 4.2−14.5 months). The authors concluded that niraparib plus pembrolizumab is a promising combination therapy in OC, but further research is needed, including biomarker analysis. There are several ongoing trials in the primary and relapsed setting to further clarify the role of the PARPi and ICI combination in OC (Table 4).

TAILORED AND TARGETED THERAPY BEYOND ANTIANGIOGENIC AGENTS, PARPI AND IMMUNOTHERAPY
Despite recent development, there is a high medical need for all OC subtypes. Currently, the most promising newer agents in the treatment of OC are PARPi and inhibitors of angiogenesis. Beyond that, there are several new agents on the horizon, which are presented in the following section.
Targeting the estrogen receptor
Although having been known for decades, the estrogen receptor (ER) might be an overlooked target. In high-grade serous ovarian cancer, ER is expressed in more than 80% of cases.43 So far, several small, non-randomized studies examined the role of endocrine therapy in relapsed OC.44 In a smaller retrospective study in 50 patients with HGSOC, the use of letrozole maintenance (n=23) was associated with a significantly prolonged recurrence-free interval, as compared with those who did not receive maintenance therapy (n=27). After 24 months, 60% of patients in the letrozole group and 38.5% of patients in the control group were recurrence-free (p=0.035).45 Furthermore, the large ongoing randomized MATAO trial is currently evaluating letrozole versus placebo in the maintenance setting in patients with newly diagnosed high-grade and low-grade serous OC.46 In the case of positive results, this trial will be practice-changing.
The recently published phase II PARAGON trial, which assessed anastrozole, a nonsteroidal aromatase inhibitor, in asymptomatic patients with relapsed ER-positive and/or progesterone receptor (PR)-positive low-grade OC, demonstrated a clinical benefit rate of around 35%.47 Within this trial, global measures of quality of life remained stable. However, in patients with high symptom and tumor burden, an aromatase inhibitor might not be the optimal therapy because of the longer response time. In addition, chemotherapy is not efficient in this second most common OC subtype.
The combination of a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor with endocrine agents might also be an effective strategy in the relapsed or even primary maintenance setting. CDK4/6 inhibitors target the cyclin D-CDK4/6 complex and suppress the activation of the retinoblastoma (Rb)-EF2 pathway, thereby interrupting G1/S cell cycle transition and, in turn, the tumor cell proliferation.48 CDK4/6 inhibitors like palbociclib, ribociclib and abemaciclib are standard of care drugs for patients with ER-positive, HER2-negative metastatic breast cancer.49–51
In 2019, the results of a phase II study investigating ribociclib and letrozole in patients with relapsed ER-positive OC or endometrial cancers were published.52,53 In this study, 40 patients with OC (n=20) and endometrial cancer (n=20) received ribociclib (400 mg on 21 days of 28-day cycle) and letrozole (2.5 mg daily). In the OC cohort, the 12-week PFS rate was 50%. Of note, 3/3 of patients with low-grade serous OC (LGSOC) achieved at least a partial response lasting for over 2 years. These combinations will be evaluated for further clinical use and is a promising target for patients with OC.
Targeting the MEK/ERK pathway
LGSOC, a relatively rare OC subtype, is associated with low chemotherapy response and common alterations in the MAP kinase pathway, most frequently in the KRAS and BRAF proteins. A large number of case series identified the high rate of KRAS and BRAF mutations in LGSOC and serous borderline tumors.54 MEK inhibitors are emerging as active agents and may well represent an effective therapeutic strategy in the near future. Recently, binimetinib, a potent MEK1/2 inhibitor with demonstrated activity across multiple cancers, was evaluated in LGSOC patients. In the large phase III MILO/ENGOT-ov11 study, 303 patients with recurrent LGSOC after ≥1 prior platinum-based chemotherapy but ≤3 prior chemotherapy lines were randomized 2:1 to receive either binimetinib or physician’s choice chemotherapy (PCC).55 The primary endpoint of the study was not met, with a median PFS of 9.1 months with binimetinib and 10.6 months with PCC (HR: 1.21 [95% CI: 0.79−1.86). However, binimetinib showed antitumor activity in LGSOC across other efficacy endpoints evaluated. Of note, in a post hoc analysis of MILO, the presence of a KRAS mutation appeared to predict benefit from binimetinib in women with LGSOC.56 Among binimetinib-treated patients, the median PFS was significantly prolonged for those with KRAS mutations versus wild-type tumors (17.7 months vs 10.8 months; p=0.006).
Another MEK inhibitor, trametinib, has been evaluated in the phase II/III NRG-GOG 0281 trial which showed that treatment with trametinib resulted in a 52% reduction in the risk of disease progression or death.57 The median PFS was 13.0 months with trametinib compared with 7.2 months for physician’s choice of SOC therapy (HR: 0.48 [95% CI: 0.36−0.64]; 1-sided p<0.0001). Median OS was longer with trametinib versus SOC, but the results did not reach statistical significance. This drug seems to be an evaluable second- or later-line option for patients with relapsed LGSOC.
Very recently, the combination of VS-6766, a RAF/MEK inhibitor, and defactinib, a FAK inhibitor, was granted FDA breakthrough therapy designation for the treatment of patients with recurrent LGSOC, irrespective of KRAS status, after at least 1 prior therapy line.58 In the ongoing phase I/II FRAME trial,59 which aimed to assess this combination therapy in patients with various advanced solid tumors. In the LGSOC cohort (n=24), the treatment led to an ORR of 52%. When stratified by KRAS status, the ORR was 70% in patients whose tumors were KRAS-positive (n=10), while it was 44% in those with wild-type KRAS (n=9).
Cell cycle inhibition beyond CDK4/6 inhibitors
In the last years, several cell cycle proteins have been discovered as promising targets in OC therapy, including the p53 mutations in HGSOC and kinases involved in G2/M checkpoints.60 In solid tumors, CHK1 inhibitors like prexasertib and ATR-inhibitors like ceralasertib and berzosertib showed encouraging results.61–64 Greater clinical benefit was achieved when combining these agents with highly effective and well-tolerated drugs such as PARPi.65,66 However, due to overlapping toxicities resulting mainly in severe hematological side effect, the safety of combination therapies with cell cycle inhibitors need to be better evaluated in ongoing clinical trials.
WEE1-kinase inhibitors like adavosertib seem to sensitize platinum-resistant OC cells to platinum-based chemotherapy. A recently published phase II study showed promising activity of adavosertib plus gemcitabine in patients with relapsed platinum-resistant OC.67 In total, 124 women (HGSOC: n=99; non-HGSOC: n=34) were randomized 2:1 to receive intravenous gemcitabine (1,000 mg/m2 on days 1, 8 and 15) with either oral adavosertib (175 mg on days 1, 2, 8, 9, 15, and 16) or placebo in 28-day cycles, until disease progression or unacceptable toxicity). At data cutoff, the median PFS was 4.6 with adavosertib and 3.0 months with placebo (HR: 0.55; p=0.015) in this heavily pre-treated group. Among HGSOC patients, the most common frequent grade ≥3 adverse events (AEs) were hematological and included neutropenia (62% with adavosertib; 30% with placebo) and thrombocytopenia (31% vs 6%).
Targeting the folate receptor-alpha
Folate receptor alpha (FRα) is overexpressed in more than 90% of OC and might be a promising target for therapy with antibody-drug conjugate (ADC).68 Mirvetuximab soravtansine (MIRV) is an ADC comprising an FRα-binding antibody, cleavable linker, and the maytansinoid DM4, a potent tubulin-targeting agent. The randomized, open-label, phase III study FORWARD I compared MIRV and investigator’s choice chemotherapy in patients with platinum-resistant epithelial OC.69 In this trial, 366 patients treated with 1−3 prior lines of therapy and whose tumors were positive for FRα expression underwent 2:1 randomization to receive either MIRV (6 mg/kg) or chemotherapy (paclitaxel, pegylated liposomal doxorubicin or topotecan). The primary endpoint of PFS was not met. The median PFS was 4.1 with MIRV versus 4.4 months with chemotherapy in the ITT population (HR: 0.981), while it was 4.8 and 3.3 months, respectively, among patients with high FRα expression (n=218) (HR: 0.693; p=0.049). In the FRα-high population, superior clinical outcomes with MIRV versus chemotherapy were observed in all secondary endpoints including ORR (24% vs 10%), CA-125 responses (53% vs 25%) and patient-reported outcome measure (PROM) (27% versus 13%). In the overall population, MIRV versus chemotherapy was associated with lower rates of treatment-related grade ≥3 (25.1% vs 44.0%), as well as AEs leading to dose reduction (19.8% vs 30.3%) and treatment discontinuation (4.5% vs 8.3%). These encouraging results have led to two subsequent studies of MIRV in patients with platinum-resistant, FRα high epithelial OC. The ongoing single-arm SORAYA study is designed to support accelerated approval, with ORR as the primary endpoint.70,71 This study enrolled only patients with high FRα expression and has a more specific diagnostic test, in comparison with FORWARD I. Furthermore, the ongoing randomized phase III MIRASOL trial comparing MIRV with chemotherapy has been designed as a confirmatory study, with PFS as the primary endpoint.72
Based on the results from the FORWARD I study, the FDA and EMA granted orphan drug designation to MIRV for the treatment of OC.73 In June 2018, the FDA granted MIRV Fast Track designation for the treatment of patients with a medium to high FRα-positive platinum-resistant OC who received 1−3 prior systemic therapies and for whom single-agent chemotherapy is appropriate as the next line of therapy. However, the final regulatory approval will be based on the findings from SORAYA.
MIRV has also been investigated in combination with bevacizumab in 66 heavily pretreated patients with FRα-positive, platinum-resistant ovarian cancer in the phase Ib FORWARD II study.74 At data cutoff, the confirmed tumor response was achieved in 26 (39%) patients, including 5 CRs and 21 PRs. The median PFS was 6.9 months. The combination was particularly active in the subset of patients (n=16) who were bevacizumab-naïve, less heavily pretreated and whose tumors exhibited medium/high FRα expression, with an ORR of 56% and the median DoR of 12 months. The median PFS was 9.9 months. AEs were mainly of ≤grade 2, with diarrhea, blurred vision, nausea and fatigue being the most common treatment-related toxicities. Altogether, 6 patients experienced pneumonitis (all grade 1−2).
CONCLUSIONS
The prognosis of patients with OC has been increased over the last decades. The current cornerstone is optimal surgical treatment and platinum-based chemotherapy. In terms of targeted therapy, the most promising agents include antiangiogenic drugs and PARPi. In the maintenance setting, bevacizumab, niraparib, olaparib and rucaparib are approved treatment modalities, which can also be used in the relapsed setting.
Immunotherapy with checkpoint inhibitors is under development. However, the first clinical results showed that single-agent activity is minor and the combination with chemotherapy led to disappointing outcomes. On the other hand, the combination of antiangiogenic drugs and PARPi provided encouraging results. There is however no clear biomarker or signature which patient most profit from the combinations. Patients with tumors expressing PD-L1 ≥5% showed minimal benefit in the first-line treatment with atezolizumab. Beyond inhibitors of angiogenesis and PARPi, there are several emerging targeted therapeutics. In the future, there will be a more tailored approach based on the molecular mechanism and changes between the different subtypes.
CONFLICT OF INTEREST
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHORS’ CONTRIBUTION
M. V. has drafted the manuscript; A. L. has critically revised the manuscript. Both authors have approved the final version of the publication.