

INTRODUCTION
Systemic amyloidosis is a rare and heterogeneous group of diseases with an incidence of about 10 patients per one million inhabitants.1 Data on the epidemiology of systemic amyloidosis in Europe are still scarce. In amyloid light-chain (AL) amyloidosis, a small plasma-cell clone and, less frequently, lymphoplasmacytic lymphoma or marginal zone lymphoma produce a toxic light chain. This light chain can form amyloid fibrils in several tissues and their deposition is the direct cause of organ dysfunction. Additionally, free light chains (FLCs) can have direct toxic effects, e.g., on cell metabolism and cardiac function.2 Organ dysfunction frequently involves the kidneys (46%), which usually leads to nephrotic syndrome, and the heart (30%), which leads to heart failure with preserved ejection fraction (HFpEF). Other organs are affected less frequently, including the liver (9%), with hepatomegaly, the gastrointestinal tract (7%), the peripheral nervous system (5%) and the soft tissues (3%).3 In contrast, localized deposition of the light chains can occur in the skin, respiratory system and the gastrointestinal and genitourinary tracts, as well as in the eyes and in the lymph nodes. This can lead to local symptoms, which are mainly treated by surgical removal of the deposits.4
Early diagnosis of systemic AL amyloidosis is important, as it not only improves overall survival (OS) and prevents organ dysfunction, but also allows a wider selection of therapies and improves the quality of life (QoL).1 On the other hand, a diagnosis that is driven by clinical symptoms or signs is often late, which severely affects both treatment options and QoL.
Several novel therapies for plasma-cell neoplasms have been introduced into clinical practice in recent years. As these therapies are directed against clonal plasma cells, they have also been studied for AL amyloidosis, thus increasing the therapeutic options for this patient population. These novel agents include proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs) and monoclonal antibodies against CD38 or the surface glycoprotein SLAMF7, both highly expressed on the clonal plasma cells. All these treatment options have been successfully used in various combinations for plasma-cell neoplasms. This makes the prompt and efficient treatment of patients with AL amyloidosis possible, thus preventing the progression of organ dysfunction.
The focus of the present review on systemic AL amyloidosis is the diagnostic work-up, aiming at an early and correct diagnosis of the disease. We also present the biomarker-based risk stratification that forms the basis of individualized treatments of patients with AL amyloidosis, along with the therapeutic options in the time of precision medicine and the monitoring of clinical responses to therapy. Diagnosis and treatment are best discussed in multidisciplinary tumor boards, to optimize disease management in this often fragile and difficult-to-treat patient group.
The Swiss Amyloidosis Network (SAN) comprises a collaboration of various experts in the field of amyloidosis, to generate and implement evidence, guide the diagnostic work-up and the treatment of patients, in order to improve the quality of care for patients with amyloidosis in Switzerland.
From pathophysiology to biomarkers
In AL amyloidosis, monoclonal gammopathy is always present and usually, a small plasma-cell clone with median bone marrow infiltration of about 10% produces the toxic light chain. Therefore, the measurable FLCs in the serum are often only slightly elevated. There is a predominance of lambda versus kappa light chains in AL amyloidosis.5 These increased FLC levels can precede symptoms of amyloidosis by over 4 years. Screening of patients with known monoclonal gammopathy of undetermined significance (MGUS) and an abnormal FLC ratio for pre-symptomatic amyloid organ involvement can be carried out using established biomarkers. The Swiss Amyloidosis Network (SAN) recommends bi-annual screening of patients with MGUS and an abnormal FLC ratio, and with smoldering myeloma.6
Even before overt heart failure and nephrotic syndrome occur, the measurement of N-terminal B-type natriuretic peptide (NT-proBNP) levels can detect cardiac amyloidosis, and albuminuria can be indicative of renal amyloidosis. These biomarkers can thus help to establish a diagnosis of AL amyloidosis prior to irreversible organ damage.7 Analysis of NT-proBNP, albuminuria and alkaline phosphatase should be performed at diagnosis of MGUS and at each follow-up visit. However, the cost-effectiveness of this approach remains an open question.
Initiation of diagnostic workup at low-level suspicion: is monoclonal gammopathy present?
In case the presence of monoclonal protein is unknown, initiation of diagnostic work-up should start at low levels of suspicion, which should usually be based on clinical signs and symptoms. The clinical signs and symptoms that indicate possible amyloid organ involvement are usually unspecific, and also multiple signs and symptoms can occur simultaneously. The recommended diagnostic work-up is summarized in Figure 1.
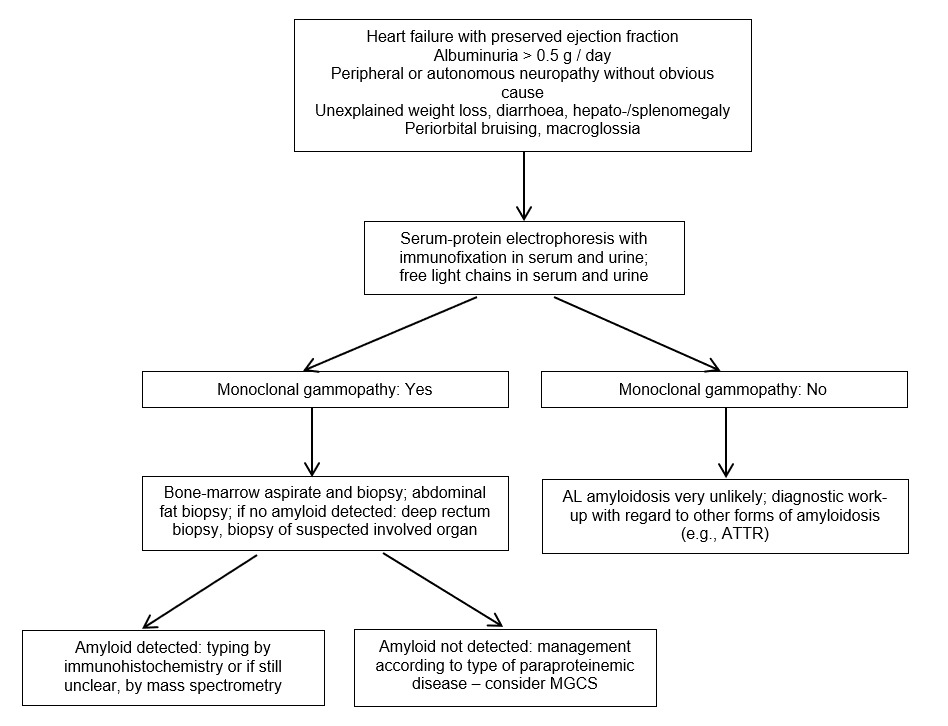
The very common symptoms of AL amyloidosis are proteinuria/albuminuria progressing to renal failure and heart failure with preserved ejection fraction, along with the increased thickness of the ventricle wall and often low voltage on the electrocardiogram. Other potential but infrequent signs of organ involvement due to amyloid fibrils include hepatomegaly with cholestasis, postural hypotension, syncope, peripheral neuropathy (as ascending, symmetric, small fibers/axonal), gastrointestinal mobility alterations, unintended weight loss, periorbital purpura (i.e., “raccoon eyes”), macroglossia and swelling of the submandibular salivary glands and the shoulder (i.e., “shoulder pad” signs).8
In symptomatic patients, immunofixation of serum and urine and measurement of FLC have to be performed to check for the presence of monoclonal gammopathy. If a monoclonal protein is present, tissue diagnostic for AL amyloidosis has to be carried out. If a monoclonal protein is absent in patients with suspected cardiac amyloidosis, a cardiac scintigraphy-based diagnostic pathway with bone tracers is recommended, to look for transthyretin (ATTR) amyloidosis.8 Molecular genetic analysis is necessary to differentiate between hereditary (rare) and wild-type (frequent) ATTR amyloidosis.
Monoclonal gammopathy of clinical significance (MGCS) has to be considered in patients with a monoclonal gammopathy experiencing unexplained symptoms especially in the nerves (“Neurologic MGCS”), the kidneys (Monoclonal gammopathy of renal significance, MGRS) and the skin (“Cutaneous MGCS”), and nearly all of the renal and dermatologic diseases are diagnosed by either renal or skin biopsies. Several of such conditions are syndromes (e.g. POEMS syndrome), therefore it is important to recognize other manifestations.
Tissue diagnosis and amyloid typing
There are several ways to perform tissue diagnosis and amyloid typing. Amyloid deposits can be found in bone marrow with a sensitivity of about 70% using Congo red staining and immunohistochemistry (IHC). Amyloid fibrils can be detected by light microscopy, where they show anomalous birefringence under polarized light after Congo red staining. Amyloid deposits can also be found in abdominal fat and/or the minor salivary glands. The combination of bone marrow and abdominal fat aspirate examinations can yield a diagnostic sensitivity of about 90% at referral centers.9 Unfortunately, these results frequently cannot be reproduced by non-referral centers. Also, abdominal fat aspirates are not suitable for IHC, which is performed almost exclusively on tissue biopsies. Therefore, in combination with bone marrow aspiration and biopsy, an abdominal fat biopsy might be the better option for correct diagnosis. With such a diagnostic approach, most patients do not need a biopsy of the organ involved, e.g., the heart or kidney. Also, the bleeding risk can be increased due to vascular amyloid deposits and possible clotting factor X deficiency, as a result of its adsorption onto amyloid fibrils.10 On this basis, if a liver biopsy is to be performed at all, it should only be performed through venous transjugular access.
As the clinical presentation of systemic AL amyloidosis and other types of systemic amyloidosis (e.g., ATTR amyloidosis) can be very similar, amyloid tissue typing with adequate technology is mandatory. In most patients, this is done with light microscopy IHC with custom-made antibodies, which can correctly classify 94% of patients. Alternatively, electron microscopy IHC with commercially available antibodies can correctly classify more than 99% of patients.11 While mass spectrometry is considered the gold standard for amyloid typing, it is not currently available in Switzerland, but it can be performed at the Amyloidosis Research and Treatment Center of the University of Pavia, Italy, or other amyloidosis centers worldwide. It should be noted that about a quarter to a third of the most elderly patients with wild-type ATTR amyloidosis have a monoclonal component, which might only be an innocent bystander unrelated to cardiac amyloidosis.12 This underlines the importance of correct amyloid typing, as AL amyloidosis is by far the most rapidly progressing type of cardiac amyloidosis, where the most clinical benefits can be obtained by an early start of anti-plasma-cell therapy.
Staging and risk stratification
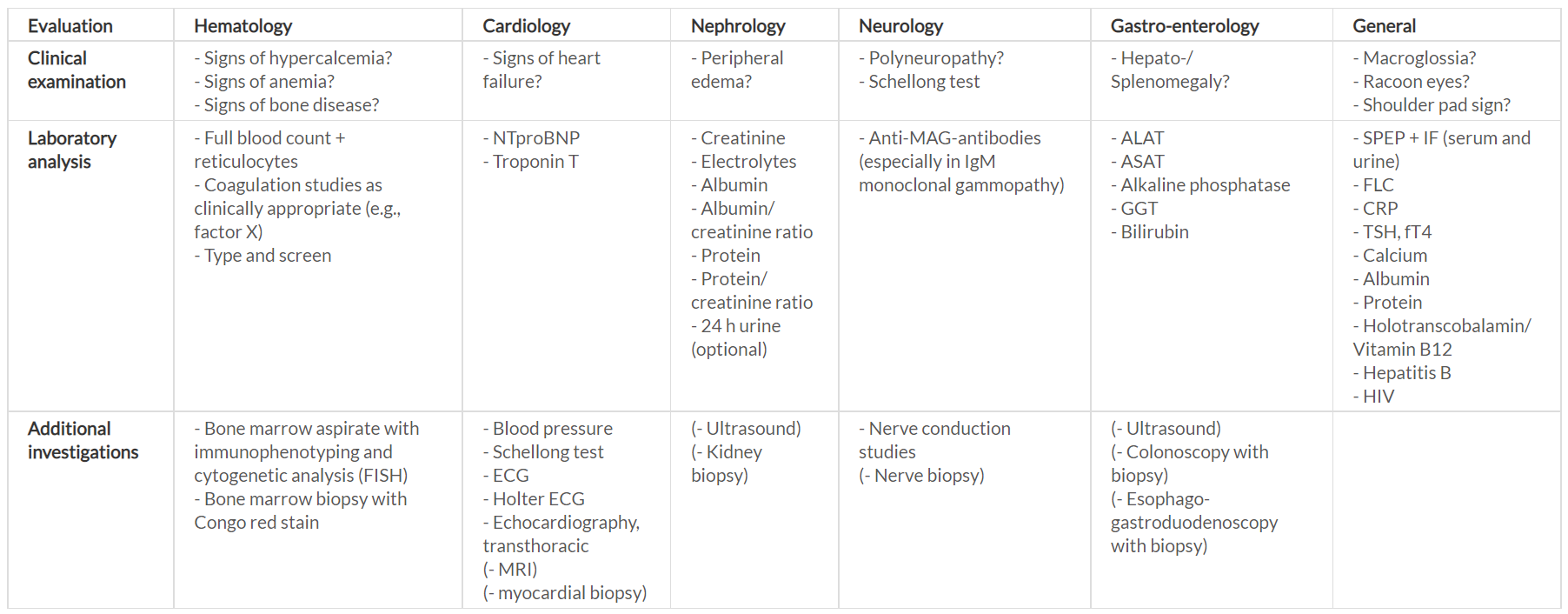
Once the diagnosis of AL amyloidosis has been established, it is necessary to assess the general health status of the patient, the biology of the disease and the extent of organ involvement, to individually design a risk-adapted therapeutic strategy. Therefore, a structured baseline evaluation is desirable, as highlighted in Table 1.

In addition to the evaluation of comorbidities, immunofixation of serum and urine, as well as determination of FLC, have to be performed. Bone-marrow examination usually reveals a small plasma-cell clone with a median plasma-cell infiltrate of about 10%. Immunophenotyping and cytogenetic analysis of the bone-marrow aspirate need to be performed at diagnosis. In particular, cytogenetic analysis is crucial in the time of precision medicine, as it can influence the choice of first-line therapy. About 50% of plasma-cell clones harbor translocation (t)(11;14), and about 20% show the gain 1q21. Instead, the high-risk aberrations such as t(4;14) and deletion 17p are infrequent, being found in <10% plasma-cell clones.13 A skeletal survey will complete the evaluation of the plasma-cell clone.
Further analyses should include at least NT-proBNP, cardiac troponin, an electrocardiogram (ECG), a Holter ECG, transthoracic echocardiography and cardiac magnetic resonance imaging (MRI). There should also be an analysis of creatinine, with the estimated glomerular filtration rate (eGFR), and albumin and protein in the urine, along with a liver function test. The severity of heart involvement and the FLC burden are the two main prognostic determinants in AL amyloidosis.
The Mayo Clinic/European staging system is based on NT-proBNP (cutoff, 332 ng/L) and troponin I (cutoff, 0.1 ng/mL), with stage I, II and III patients having 0, 1 or 2 markers, respectively, above the cutoffs.14,15 Patients with very high NT-proBNP (>8,500 ng/L) have advanced cardiac involvement (stage IIIb) with particularly poor outcomes. The revised Mayo Clinic staging system is based on NT-proBNP (cutoff, 1,800 ng/L), troponin I (cutoff, 0.07 ng/mL) and the difference between the involved and uninvolved serum-free light chains (dFLC; cutoff, 180 mg/L), with stage I, II, III and IV disease in patients showing 0, 1, 2 or 3 markers, respectively, above the cutoffs.1,16 In patients with predominant renal involvement, a distinct renal staging system has been developed that incorporates proteinuria and eGFR.17
Accurate risk stratification of patients with AL amyloidosis is a prerequisite for an individualized treatment strategy. Low-risk patients are usually eligible for autologous stem-cell transplantation (ASCT), whereas intermediate- and high-risk patients are – at least initially – ineligible for ASCT.8 The selection criteria that are used to determine the risk groups of patients with AL amyloidosis are summarized in Table 2.

Predictive markers in the time of precision medicine
Cytogenetic analysis of bone marrow with interphase fluorescence in situ hybridization (iFISH) and/or array-based comparative genomic hybridization (aCGH) is recommended to characterize the amyloid clone and to obtain information on the best choice of therapy. Patients with plasma-cell clones that harbor t(11;14) have a worse outcome with bortezomib and IMiDs,18 while gain 1q21 is an independent adverse prognostic factor in AL amyloidosis patients treated with melphalan.19 The treatment choice, however, is not only influenced by the genetic markers but also depends on access to drugs, the possibility to participate in clinical trials and the preference of the patient. The early improvement of organ dysfunction is possible, which can allow more aggressive treatments, and make the patients eligible for ASCT, for those who were ineligible initially.
Response assessment
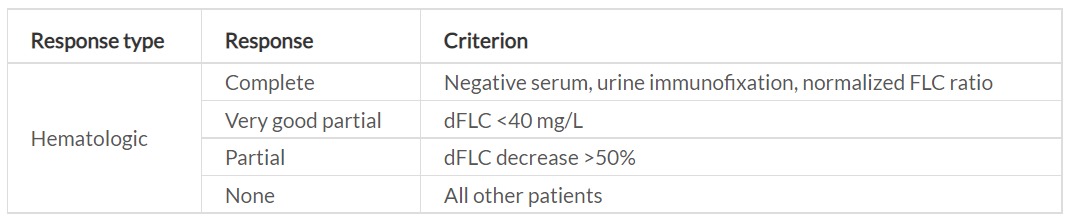
The response criteria have been validated by the International Society of Amyloidosis (ISA) in independent populations based on survival outcomes, for a definition of the hematologic responses and organ responses, as indicated in Table 3.

The hematologic response measures the effect of anti-plasma-cell therapies and is divided into the four response categories of complete response (CR), very good partial response (VGPR), partial response (PR) and no response (NR), as based on the changes in dFLC.20 The deeper the response, the better the outcome, and at least a VGPR is the goal of any chosen treatment regimen, defines by an OS rate of 80−90% after 3 years. Organ response is the ultimate goal of the therapy, although this is usually a more long-term outcome after obtaining a hematologic CR, as it can take as long as 9.4 months for the heart, 6 months for the kidneys and 6.1 months for the liver.21
The validated composite hematologic plus organ response (CHOR) model, which combines hematologic response and organ response, classifies patients into one of two CHOR groups, according to a score based on the hematologic and organ response criteria: CHOR group 1, scores 0−3; CHOR group 2, scores 4−5. This model has been rarely used in studies and clinical practice to date, as the hematologic and organ responses are still generally considered separately.
The continuing effectiveness of any therapy should be verified frequently. As the FLCs have a half-life of hours, the hematologic response should become apparent shortly after treatment initiation. The treatment regimen should also be changed rapidly if at least a PR (dFLC <50%) after two to three cycles of therapy has not been achieved.
Structured assessment and documentation of the hematologic and organ responses at least every 3 months is recommended by the Swiss Amyloidosis Network. During induction therapy, the hematologic response should be determined at least on day 1 of every therapy cycle and at the end of every treatment. Novel definitions of deep hematologic responses are being investigated that include determination of minimal residual disease (MRD) using multiparameter flow cytometry.22
THERAPY FOR AL AMYLOIDOSIS
The major challenges in the selection of treatment regimen for patients with AL amyloidosis include balancing of the expected efficacy, the characteristics of the plasma-cell clone and the access to drugs on one side, and severity of organ involvement and specific contraindications (e.g., neuropathy, renal failure, patient frailty) on the other side, while also considering the preferences of the patient. The treatment should always be based on an individualized, risk-adapted and response-tailored approach.
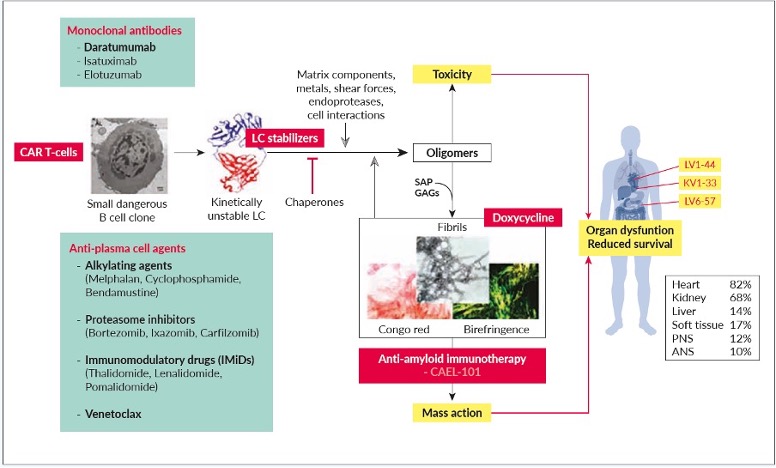
There are potentially several treatment targets in AL amyloidosis, as summarized in Figure 2.23 The plasma-cell clone is the most obvious of these and represents the cornerstone of any therapeutic approach. The aim of the therapy is suppression of the production of light chains, to restore organ function, especially cardiac function. This can be achieved with classical anti-plasma-cell agents, such as alkylating agents, proteasome inhibitors, IMiDs, monoclonal antibodies and/or venetoclax.

The alkylating agents such as melphalan, cyclophosphamide and bendamustine can be very effective for the treatment of AL amyloidosis. Oral melphalan in combination with dexamethasone can yield dose-dependent hematologic response rates of 51−76%, with minimal toxicity.24 Indeed, this combination was previously considered as standard therapy for patients who were ineligible for ASCT. It is most suitable for elderly patients with revised Mayo stage I/II who have no need to start treatment immediately. Cyclophosphamide is usually part of the combination with bortezomib and dexamethasone (CyBorD), and oral once-weekly treatment is the typical mode of application. Bendamustine with or without rituximab is usually used for patients with AL amyloidosis with a monoclonal IgM protein when the pathological cell clone is usually lymphatic in origin (e.g., IgM-MGUS, Waldenström macroglobulinemia, marginal zone lymphoma, chronic lymphocytic leukemia). It is reasonably well tolerated and effective, with hematologic response rates of >50%.25
Monoclonal antibodies alone and in combination with alkylating agents and other monoclonal antibodies are also used, while CAR T cells, small-molecule kinetic light chain stabilizers suppressing aggregation and doxycycline are potential treatment options. Doxycycline is an antibiotic that has shown fibril-stabilizing effects in a transgenic mouse model.26 A phase III study is currently comparing doxycycline with standard supportive therapy in newly diagnosed patients who are undergoing bortezomib-based induction therapy. Anti-amyloid immunotherapies that target the amyloid deposits are a further potentially attractive treatment option, although the only anti-amyloid-fibril antibody that is still under study is CAEL-101, as the trials with other immunotherapies that target amyloid deposits have not been successful.
Therapy is indicated for all patients with verified systemic AL amyloidosis, although the optimal endpoint of treatment remains controversial. The ultimate goal of each therapy is an early and profound reduction of amyloid light chains, as this is associated with improvements in organ function and patient QoL, and with prolonged progression-free survival (PFS) and OS.27
Although pathological plasma-cell clone is sensitive to current treatment strategies, anti-plasma-cell drugs and their combinations are often associated with poor tolerability and increased morbidity and mortality, especially at the treatment initiation. This explains the need for either dose adjustments that start with low doses and titrate the dose up, or for combinations of drugs to be sequentially introduced during the course of the therapy.
Symptom management is also an integral part of any therapy. This especially involves salt restriction and diuretics in case of heart failure with preserved ejection fraction (HFpEF). In patients with recurrent arrhythmias and syncope, implantation of a pacemaker should be discussed, whereas the use of implantable defibrillators remains controversial. For neuropathic pain, gabapentin or pregabalin are the therapeutic options. To ensure sufficient caloric intake, nutritional support after nutritional status assessment is important, as unintended weight loss and wasting are frequent problems in AL amyloidosis. Last but not least, drugs that control bowel dysfunction, as well as anti-infective strategies, are important parts of the accompanying symptom management.
Organ transplantation is rarely performed, although it can be considered in patients with irreversible, end-stage organ dysfunction despite hematologic response. In young patients with isolated severe cardiac involvement and dysfunction, a treatment strategy can include cardiac transplantation followed by effective chemotherapy.
First-line therapy
First-line therapy in low-risk patients and the role of autologous stem cell transplantation
Low-risk patients are candidates for ASCT unless comorbidities or patient’s preference prevents consideration of this therapy. Bortezomib-based induction therapy independently increases PFS and should therefore be standard-of-care. This applies not only to patients with pathological plasma-cell clone infiltration >10%, unless there are contraindications to bortezomib, such as polyneuropathy or pulmonary fibrosis.28 Induction therapy allows an immediate start to therapy and therefore rapid reduction of the amyloid light chain, and it reveals valuable information with regard to the physical fitness of the patient prior to ASCT. Induction therapy without the inclusion of melphalan (e.g., a combination of cyclophosphamide, bortezomib dexamethasone) is clearly preferred, to reduce stem-cell toxicity and to make stem-cell collection prior to ASCT possible. It is also important to note that bortezomib-based induction therapy alone can lead to durable hematologic CR and organ response in some patients, with no need to proceed to ASCT. Such a sequential response-driven bortezomib-based therapy with the offer of ASCT only for the patients with unsatisfactory responses is associated with low treatment-related mortality (<1%) and a 5-year OS of >80%.29
High-dose therapy with melphalan should be performed at a dose of 200 mg/m2. Lowering the dose of melphalan to increase the proportion of patients eligible for ASCT does not reduce toxicity and has a negative impact on the response rate. Post-transplantation bortezomib-based therapy increases the rate of hematologic CR and extends PFS in patients with less than VGPR after ASCT, and should thus be considered in these patients.30
First-line therapy in intermediate-risk and high-risk patients
The introduction of proteasome inhibition into clinical practice has had a major impact on the treatment of patients with AL amyloidosis, as amyloidogenic plasma cells depend on their proteasomes and are exquisitely sensitive to proteasome inhibition. Therefore, the CyBorD combination has for many years been the standard-of-care for patients with AL amyloidosis who are not initially eligible for ASCT.31 In patients with cardiac involvement, bortezomib-containing therapies can worsen orthostatic dysregulation, and therefore a starting dose of bortezomib 1 mg/m2 once weekly is advisable, with dose increase in case of good tolerability. CyBorD is the preferred therapy in patients with potentially reversible contraindications to ASCT, as they might become transplant candidates if they attain organ response after first-line therapy. For patients with renal failure with an eGFR <30 mL/min/1.73 m2, CyBorD is the treatment of choice. This combination therapy is less effective in patients with clonal plasma cells that harbor t(11;14).
More recently, a phase III trial compared the combination of bortezomib, melphalan and dexamethasone (BMDex) with MDex in 109 patients, with the primary endpoint of hematologic response at 3 months.32 Hematologic response was significantly more frequent with BMDex than MDex (79% vs 52%). BMDex also significantly prolonged PFS and OS, with a 50% decrease in mortality rate. However, the cardiac and renal responses were not significantly different, and grade 3 and 4 adverse events were more common with BMDex, such as cytopenia, polyneuropathy, and heart failure. Severe chronic renal failure was an exclusion criterion, and in these patients, cyclophosphamide might be preferred over melphalan. Although there were no cytogenetic data available for this study, it was hypothesized that the combination of BMDex can overcome the effects of both t(11;14) and gain 1(q21).
The combination of oral MDex or IMiD-based regimens such as lenalidomide, melphalan and dexamethasone (LMDex) are still valuable therapeutic options in patients with the above-described contraindications to bortezomib. The PI, carfilzomib might be an option in these patients, especially when peripheral neuropathy is present, although it should not be used for patients with relevant heart involvement. Venetoclax is a selective B-cell lymphoma 2 inhibitor, which might be an attractive option for patients with t(11;14), although data to define the role of venetoclax as first-line therapy for patients with AL amyloidosis are so far lacking.
According to recently published data, the combination therapy of the monoclonal anti-CD38 antibody, daratumumab with CyBorD will become the new standard-of-care.33,34 The ANDROMEDA trial included 388 patients, with the primary endpoint of overall hematologic CR rate. Treatment with six 28-day cycles of CyBorD was compared with subcutaneous daratumumab plus CyBorD. In view of the results from this trial, the combination therapy of subcutaneous daratumumab-CyBorD has been approved by the US Food and Drug Administration (FDA) and by Swissmedic. Daratumumab at a fix dose of 1,800 mg was given weekly for the first two cycles, every 2 weeks in cycles 3 to 6, and then monthly, until major organ deterioration (MOD)-PFS or a maximum of 24 cycles. The primary endpoint was reached in 53% of patients receiving daratumumab-CyBorD and 18% of patients receiving CyBorD alone (odds ratio, 5.1 [95% CI: 3.2–8.2]; p <0.0001). The median time to achieve at least VGPR was 17 days with daratumumab-CyBorD and 25 days with CyBorD. There was also a deeper and more rapid hematologic response with daratumumab-CyBorD. Daratumumab commonly increases the rate of infections, including pneumonia, and can cause clinically relevant hypogammaglobulinemia. In patients with infections and hypogammaglobulinemia, immunoglobulin substitution should be established, and vaccination against invasive pneumococcal disease and influenza must be given prior to treatment initiation.32 It should also be noted that fatal cardiac adverse reactions can occur in about 10% of patients and that those with Mayo cardiac stage IIIa might be at greater risk. Therefore, it is highly recommended to monitor these patients more frequently for cardiac adverse reactions and to administer supportive care as appropriate. Again, a multidisciplinary approach to prevent or treat cardiac adverse events is warranted.
In both of the phase III trials that used either BMDex or subcutaneous daratumumab with CyBorD, patients with Mayo cardiac stage IIIb were excluded. This is a very difficult-to-treat patient population, and there are no data from randomized controlled trials on their optimal therapeutic management. This emphasizes the need for early diagnosis in patients with AL amyloidosis, to provide them with effective therapy with an acceptable safety profile. In particular, for patients with t(11;14), the combination of the monoclonal antibody daratumumab with either BMDex or venetoclax should be further investigated.
Therapy of relapsed/refractory disease
The choice of treatment in the relapsed/refractory setting depends on numerous factors, such as efficacy, quality of response, tolerability and safety of the first-line treatment, along with the time interval to the next proposed treatment, any specific contraindications and patient preference. There are currently no approved treatment regimens in the relapsed/ refractory setting.
If the aim of the first-line therapy is not achieved, as the suppression of light-chain production to restore organ function, the patients should receive second-line treatment as early as possible. On the other hand, if the patients do achieve the initial aim and then disease relapse occurs, there is no consensus on when treatment should be started. There is, however, agreement that the start of further treatment should not await organ progression. Indeed, even small increases in pathological light-chain levels should initiate rescue therapy, as these can precede organ progression by several months.
There are currently no data to support a second ASCT for patients with AL amyloidosis, which is different from the situation with plasma-cell myeloma. IMiDs are usually provided for rescue therapy. Lenalidomide and pomalidomide are typically given with corticosteroids, a combination that has shown activity in a pooled analysis, where 39% of patients achieved VGPR or CR.35 Treatment with IMiDs interferes with the assessment of cardiac response, as it increases NT-proBNP, while in patients with proteinuria, increases in serum creatinine have been reported. Initial dose reductions can help to increase tolerability, with lenalidomide of 5–15 mg and pomalidomide of 2–3 mg on days 1−21 of 28-day cycles.
Ixazomib in combination with dexamethasone (IDex) versus the physician’s choice (dexamethasone, melphalan-dexamethasone, cyclophosphamide-dexamethasone, thalidomide-dexamethasone and lenalidomide-dexamethasone) was assessed in the phase III trial TOURMALINE-AL1 trial enrolling 168 patients. Nearly half of them had received prior bortezomib. The primary endpoint of the study was a hematologic overall response, but this was not met (53% vs 51%); however, for the IDex group, both the CR rate (26% vs 18%) and the PFS were improved.36
There have been numerous retrospective and prospective trials with daratumumab, either as monotherapy or in combinations.37,38 Hematologic response can be achieved in around 73% to 76% of patients. In most cases, this occurs after a single infusion and is accompanied by high rates of cardiac and renal responses. Patients with nephrotic range proteinuria can show shorter event-free survival. It remains to be seen to what extent hematologic and organ response rates can be achieved with daratumumab as first-line therapy and in the relapsed disease, either alone or in combination(s). The combination of daratumumab with IMiDs might be particularly interesting in this setting.
CONCLUSIONS
In recent years, significant progress has been made in terms of diagnosis, risk-adapted treatment and response assessment for patients with AL amyloidosis, which have mainly been driven by work carried out in large research and treatment centers. It is now possible to make pre-symptomatic diagnoses using biomarkers, to individualize treatment design according to disease-associated risk, to use several mainly anti-plasma-cell drugs for treatments that have been studied in multinational trials and to reliably assess responses with validated criteria.
However, despite all this progress, major challenges remain. It is crucial to make the diagnosis early to prevent late stages of the disease, due to the restricted therapeutic options and the severe effects on patient QoL. Pre-symptomatic diagnosis is possible for patients with known monoclonal gammopathy and diagnostic analysis with regard to paraproteinemia should be performed at low levels of clinical suspicion. There remains no defined standard-of-care for high-risk patients, especially for those with advanced cardiac disease. Hematologic progression has to be defined and validated, and new sensitive techniques also need to be approved with regard to their prognostic value and therapeutic strategy, such as monitoring for minimal residual disease (MRD) with next-generation flow cytometry. The time from hematologic response to organ response can be several months, even with efficient anti-plasma-cell therapy, which is a major challenge for patients and caregivers. In this respect, in addition to anti-plasma-cell agents, drugs with treatment targets other than suppression of light-chain production might be used, such as anti-amyloid antibodies. These combinations will hopefully secure progress against this disease in the future and will thus allow precision medicine to be even more appreciated by our patients.
Take-Home Messages
-
Pre-symptomatic diagnosis of amyloid light-chain (AL) amyloidosis is possible, and it is necessary to allow the choice of efficacious therapy and preserve patient quality of life.
-
Diagnostic work-up should be started at a low level of clinical suspicion of amyloidosis, with initial monitoring for monoclonal gammopathy.
-
Tissue diagnosis and amyloid typing is crucial to differentiate between the several forms of amyloid, which has prognostic and therapeutic consequences.
-
There are numerous treatment targets and treatment options for patients with AL amyloidosis, most of which are aimed at the suppression of light-chain production, to restore organ function.
-
Response to therapy should be monitored according to hematologic and organ responses, with the therapy changed as soon as possible if the treatment goals are not achieved.
Conflict of Interest (COI)
AR: Fees from Janssen relevant for this article; fees from Amgen, AOP OrPha Swiss, Celgene/BMS, Novartis, Takeda unrelated to this article. There is no role of fees with regard to the manuscript.
RS: Fees from Alnylam, Janssen and Pfizer relevant for this article; fees from Mundipharma, Takeda unrelated to this article. There is no role of fees with regard to the manuscript.
Authors’ Contributions
Drafting of the manuscript: AR.
Critical revision of the manuscript: RS.