


BACKGROUND
Pancreatic acinar cell carcinoma (ACC) is a rare tumor, accounting for about 1% of all pancreatic cancers.1 It is more common in men than women (ratio 3.6:1) and is most frequently located in the pancreatic head.2 Patients with ACC clinically present with non-specific symptoms such as weight loss, abdominal pain, vomiting, nausea, and jaundice.2,3 Although being an aggressive neoplasm, ACC has, as compared with pancreatic ductal cell adenocarcinoma (PDAC), a more indolent course and improved overall survival (OS), with the median OS ranging from 18 to 47 months.4,4
Because of the very low disease incidence, there are no large series of patients with AAC, and only information from small case series and case reports are available. Although there is currently no gold standard of ACC treatment, aggressive surgical resection with negative margins has been associated with improved long-term survival.5 ACCs are usually considered chemoresponsive to the agents that have activity against PDACs, but the therapeutic efficacy of chemotherapy in ACC has not yet been studied in controlled, prospective studies.6,7 As a result, there are no definitive guidelines for treating advanced ACC. However, multidisciplinary treatment for AAC of the pancreas should be considered in the advanced setting due to the rarity of the disease and the lack of high-level therapeutic data.
PRESENTATION OF CASE
In October 2018, a 69-year-old man with a medical history of alcoholic steatohepatitis, epilepsy due to alcoholism and sleep deprivation, arterial hypertension, and cervical radiculopathy with cervical spondylosis was admitted to the emergency room with the appearance of jaundice, fever and diarrhea. The Eastern Cooperative Oncology Group (ECOG) performance status (PS) of the patient was 1, and the physical examination revealed jaundice, but otherwise unremarkable. The laboratory data was as follows: serum total bilirubin, 134.7 µmol/l (normal range <21 µmol/l); gamma-glutamyl transpeptidase, 2,043 U/L (normal range 10−71 U/L); alkaline phosphatase (ALP), 373 U/L (normal range 40−129 U/L); serum aspartate aminotransferase (AST), 175 U/L; alanine aminotransferase (ALT), 173 U/L (normal range 10−50 U/L). Other laboratory data were within normal limits.
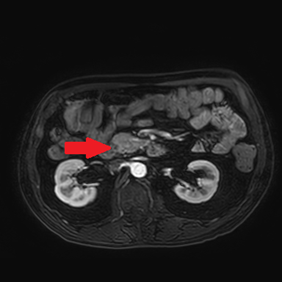
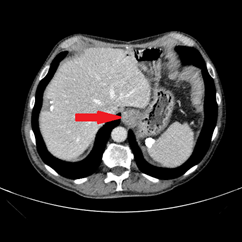
An abdominal ultrasound showed no signs of cholecystitis. A computed tomography (CT) scan revealed dilatation of the intrahepatic and extrahepatic bile duct as well as enlargement of the pancreatic head with ill-defined margins. Following hospitalization, an endoscopic sphincterotomy and endoscopic retrograde biliary drainage with 8.5 fr × 9 mm plastic stent were performed. The brush cytology of the common bile duct performed during the ERCP (endoscopic retrograde cholangiopancreatography) was positive for adenocarcinoma type malignant cells whose cytomorphological and immunophenotypic aspects were compatible with biliopancreatic primary. A magnetic resonance imaging (MRI) scan showed a 2 cm hypointense mass in the pancreatic head, with no signs of invasion in the main blood vessels (Figure 1).
After a multidisciplinary tumor board discussion, the tumor was considered resectable, and surgical treatment was recommended.
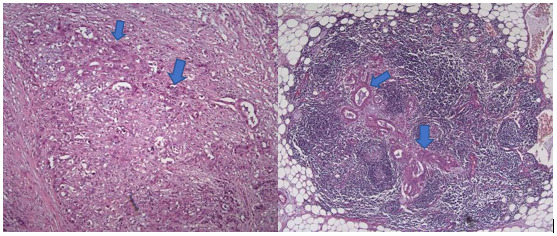
In November 2018, a pancreaticoduodenectomy (Whipple procedure) was performed. Complications after surgery were a hemoperitoneum, which was managed surgically, and peripancreatic fluid collection, which was treated with antibiotics. The postoperative histological examination of the tumor sample confirmed a moderately differentiated (G2) acinar adenocarcinoma of the pancreatic head, diameter 2.3 cm (pT2), with metastases in 1 out of 12 peripancreatic lymph nodes (N1) and the presence of perineural invasion (Pn1), without angioinvasion (V0), and negative resection margins (R0) (Figure 2).
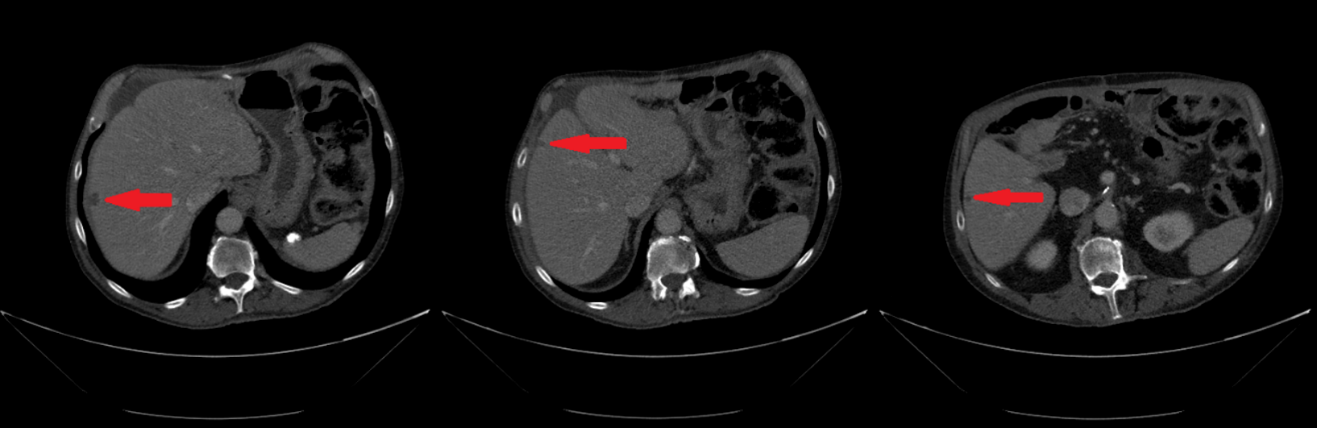
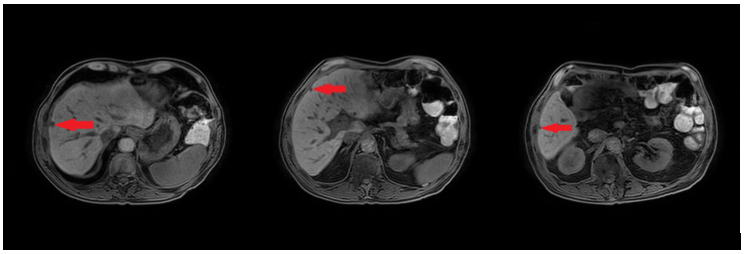
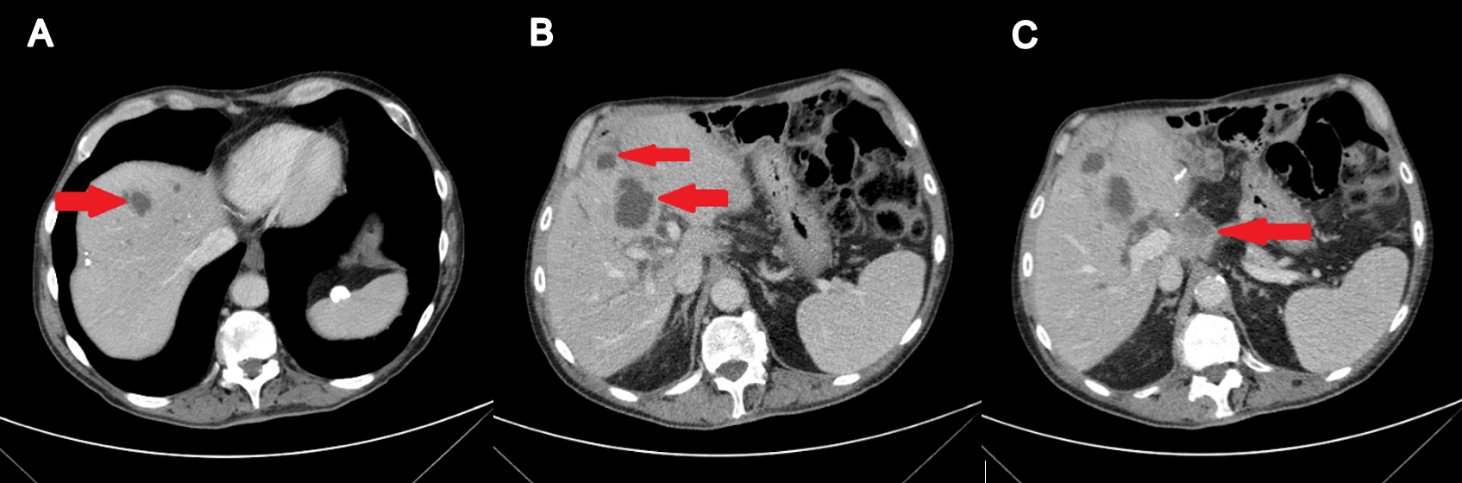
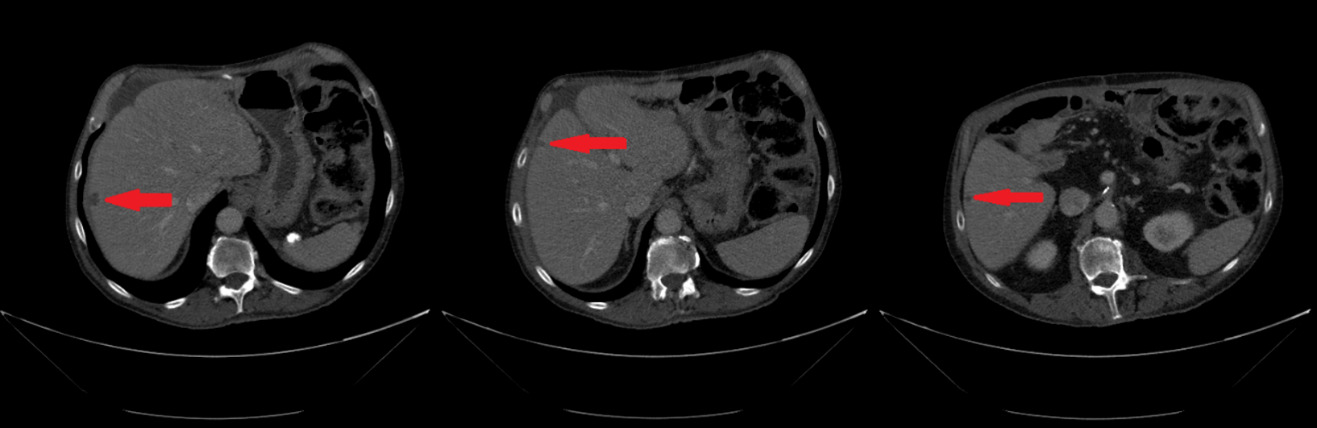
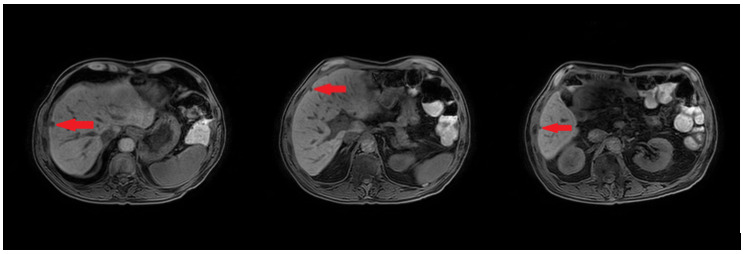
In February 2019, adjuvant chemotherapy with folinic acid, fluorouracil, irinotecan, and oxaliplatin (modified FOLFIRINOX) was initiated. About 2 months later, the serum laboratory test showed an increase in the level of the tumor marker, carbohydrate antigen (CA19.9) during chemotherapy (from 48.5 kU/l to 465 kU/l; normal range: <27.0 kU/L). In addition, a CT scan showed the appearance of 3 subcapsular lesions suspected to be metastases in the liver segment II and between liver segments V and VI (Figure 3). Thereafter, the patient received first-line palliative chemotherapy with nab-paclitaxel plus gemcitabine. A restaging with an abdominal MRI scan after 6 courses of chemotherapy showed a partial response manifested as a decrease in the size of the liver metastasis. No new lesions were detected. Furthermore, there was a progressive decrease of the CA19.9 level (from 465 kU/l to 55.2 kU/l) (Figure 4). Taking into consideration that the long-term survival of patients with AAC is usually improved as compared with PDAC patients and that radical surgery has been associated with favorable long-term survival, along with the patient’s motivation, we discussed the case in a multidisciplinary tumor board and decided for surgery of the liver metastasis.
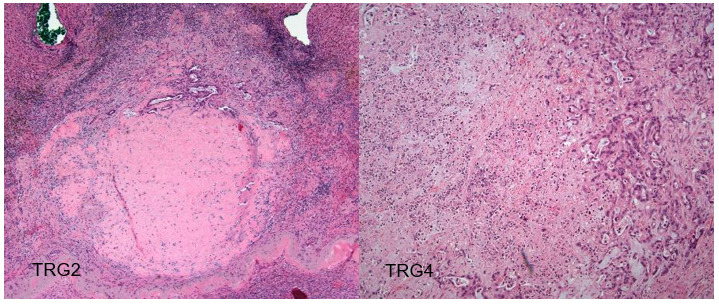
In October 2019, the patient underwent explorative laparotomy wedge resections of the IV, VI, and VII liver segments. The histological analysis showed complete resection of 3 metastases with negative margins, with tumor regression grading (TRG) scores of 2 and 4 according to the Rubbia-Brandt scoring system (Figure 5).8
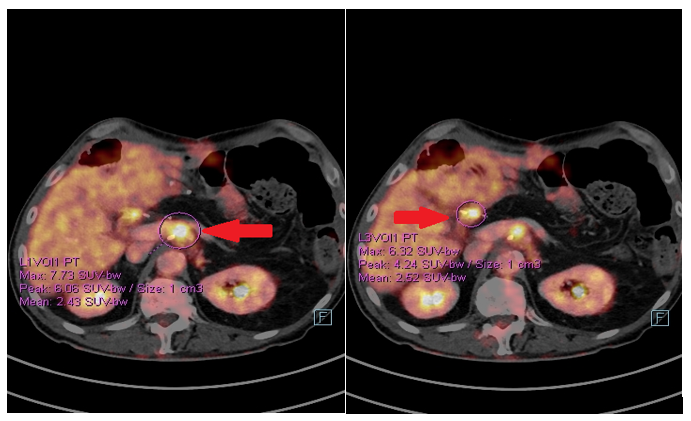
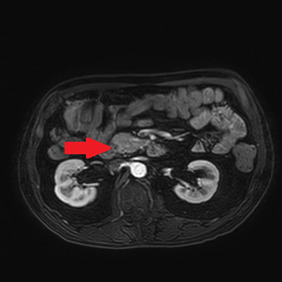
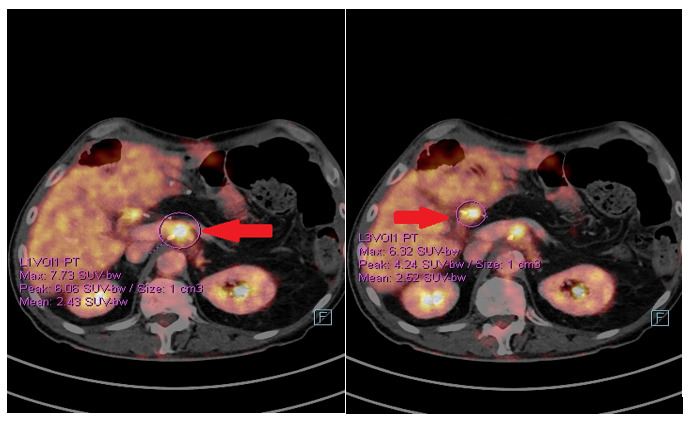
The postoperative course was uneventful and the patient started a follow-up. In January 2020, a positron emission tomography (PET)/CT scan showed the appearance of pathological tissue around the celiac trunk and in the liver hilum, which was compatible with relapse (Figure 6). Furthermore, an increase in the CA19.9 level (157 kU/l; normal range <27.0 kU/L) was detected. From 5 February 2020, the patient received rechallenge chemotherapy with nab-paclitaxel plus gemcitabine. After 4 courses of chemotherapy, he achieved a partial response manifested as a mild dimensional reduction of the solid tissue around the celiac trunk and a reduction of CA19.9 (from 447 kU/L to 115 kU/L; normal range <27.0 kU/L). However, at the end of July 2020, a rise in the CA19.9 level (from 115 kU/L to 269 kU/L) was observed, and a subsequent CT scan showed a minimal radiological progression due to an increase of the periceliac lesion (Figure 7). An ECOG PS of 0 was assessed. Thereafter, at the end of August 2020, the patient started second-line chemotherapy with liposomal irinotecan plus fluorouracil (5-FU). In the meantime, to identify a potential therapeutic target therapy, genomic profiling using next-generation DNA sequencing (NGS) and microsatellite instability analysis were performed. NGS identified the presence of KRAS G12D (exon 2) and TP53 R2495 (exon 7) mutations, while the microsatellite instability status was stable (MSS). Therefore, the possibility of targeted therapy or immunotherapy was excluded.
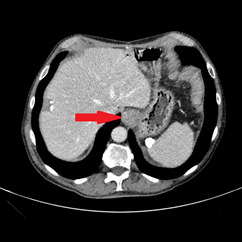
After 4 courses of therapy, a CT scan showed new lesions in the hepatic segments VIII and IV and a further increase of the periceliac pathological tissue resulting in a dilatation of the intrahepatic bile duct (Figure 8). Furthermore, a sharp increase in the CA19.9 level (1,997 kU/l) was observed. The patient also developed a cholestatic-cytolytic hepatopathy with increased levels of bilirubin (42 mmol/l; normal range <21 µmol/l), ALP (1,509 IU/L; normal range 40−129 U/L), AST (385 IU/L), and ALT (502 IU/L; normal range 10−50 U/L). The ECOG PS was 1. Since there were no other treatment options, and no phase I studies were available, we addressed the patient to the best support care (BSC).

About 1 month later, the patient was admitted to our hospital due to acute liver failure, metabolic encephalopathy, and likely hepato-renal syndrome, with subsequent death.
DISCUSSION
Although the prognosis of pancreatic ACC is generally poor, ACC is associated with favorable clinical outcomes at all stages, as compared with ductal adenocarcinoma (PDAC). The long-term survival for patients with ACC is significantly better when compared to the long-term survival of patients with PDAC (Table 1). In fact, the 5-year survival rates were 17.2% for ACC versus 2.8% for PDAC at the metastatic stage, while the median OS was 25 months versus 3 months, respectively, in locally advanced, unresectable cases.9,10 Also demographic factors and outcomes are different in the two histological types. Patients with ACC are younger, are more likely to have the resectable disease and to undergo potentially curative resection, as compared with PDAC patients.

There are only limited data on the efficacy of chemotherapy in the ACC setting, as non-ductal histology is usually excluded from the main clinical trials.11 In the absence of prospective clinical data to guide therapy, treatment strategies frequently incorporate chemotherapy agents with antitumor activity in patients with PDAC. In our case, due to the local expansion and the good condition of the patient, we proposed modified FOLFIRINOX, which is the best available adjuvant option, analogous with the ductal histology.11 In addition, Hashimoto et al. (2017) suggested that modified FOLFIRINOX was safe and effective in the treatment of pancreatic ACC.12 Also in treating the advanced disease, we adopted the chemotherapy regimens approved for ductal adenocarcinoma, which included gemcitabine plus nab-paclitaxel in the first-line, and liposomal irinotecan plus 5-fluorouracil in the refractory setting.13,14
Our patient experienced a good response with gemcitabine plus nab-paclitaxel, which gave him the opportunity to be considered for a maximal approach, including liver surgery. Taking into consideration that surgical resection for ACC drastically improves survival and that ACC has a better prognosis than ductal adenocarcinoma, aggressive surgery for the metastatic disease may be justified even though no precedent exists.1,4,15,16 In the present case, we thoroughly informed the patient on the absence of evidence to recommend our decision process, but the good response to the systemic treatment, as well as the option of a treatment holiday without disease progression, motivated the surgical intervention.
Interestingly, in our patient, the pathological response to chemotherapy was non-univocal, as a different pattern of regression was observed in the metastases, probably as a result of intratumor heterogeneity. Unfortunately, the tumor recurred at the first radiological assessment, at 3 months from the intervention, showing more aggressive behavior. The patient finally died at 1 year from the tumor relapse after liver surgery.
It is well-known that the genomic landscape of ACC is distinct from other pancreatic tumors.17–19 Typical genetic alterations observed in PDAC are normally not detected in ACC or occur rarely, such as mutations in the KRAS (~2% ACCs vs. >90% PDACs), TP53 (9–23% vs 75%), CDKN2A (14% vs 90%), SMAD4 (14–19% vs 55%) genes. This demonstrates that ACCs constitute an entity different from ductal adenocarcinoma or endocrine tumors. As in our patient KRAS and P53 mutations were found by using NGS, and we can speculate that these molecular alterations conferred a more aggressive phenotype.
The most frequent molecular alteration in ACC is the allelic loss on chromosome 11p, which is present in 50% of patients, while the second most common is the mutation in the APC/β-catenin pathway present in 23.5% of patients (Table 1). In contrast, there is no evidence for the involvement of p53 or DPC4 genes.20,21 Genomic profiling identified recurrent rearrangements involving BRAF and RAF1 (CRAF) in approximately 23% of ACC.22 The fusion SND1-BRAF activates the MAPK pathway, thus resulting in sensitivity to the MEK inhibitor, trametinib. ACC lacking RAF rearrangements are enriched for genomic alterations, which cause inactivation of DNA repair genes (45%) and result in sensitivity to platinum-based therapies and PARP inhibitors. Microsatellite instability (MSI) is described only in 5% of cases.
TAKE-HOME MESSAGES
-
Acinar cell carcinoma (ACC) of the pancreas is a relatively rare disease, accounting for about 1% of all pancreatic neoplasms, with limited clinical data and no consensus on a gold standard of treatment.
-
The 5‐year survival rate of ACC after surgical resection is better than that of pancreatic ductal adenocarcinoma (PDAC).
-
Since ACC usually shows less malignant behavior than that of PDAC, surgical resection could be an option even for metastatic lesions, in case the number and extent of metastases are limited.
-
In general, the typical abnormalities found in patients with ductal adenocarcinomas, including mutations in KRAS and TP53 genes, are very rare in ACC patients. These mutations are, however, associated with a more aggressive phenotype of ACC, similar to PDAC.
Informed Consent
General written consent was obtained from the patient for the publication of this case report and any accompanying images.
COI
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
All authors contributed to and approved the final manuscript.