INTRODUCTION
EPIDEMIOLOGY
MDS represents a heterogeneous group of clonal disorders of hematopoietic stem and progenitor cells (HSPCs).1 It is characterized by cytopenia, dysplasia in one of the major myeloid cell lineages and ineffective hematopoiesis; in addition, it harbors a variable risk of transformation into secondary acute myeloid leukemia (AML).2 The median age at diagnosis is above 70 years, with a rare presentation before the age of 50 years with the exception of therapy-related MDS or, especially, myeloid neoplasm with germ-line predispositions.3 The age-standardized incidence rates range from 3−5 cases per 100,000 patient-years and the prevalence of around 20 patients per 100,000 individuals in Western countries. MDS is predominant in males with the exception of MDS with del (5q).4–7 There is a substantial increase in the age-specific incidence-rate in patients >75 years, which is in accordance with the general increase of cancer with aging. We observe a steady increase of MDS incident cases that is related to the aging of the population, improved cancer survivorship and higher diagnostic sensitivity in identifying clonality in cases with unclear cytopenia.4 It stands to reason, that MDS is an emerging and challenging hematological malignancy with a relevant impact on healthcare resources, and as such, it requires a structured and evidence-based management approach.
PATHOPHYSIOLOGY
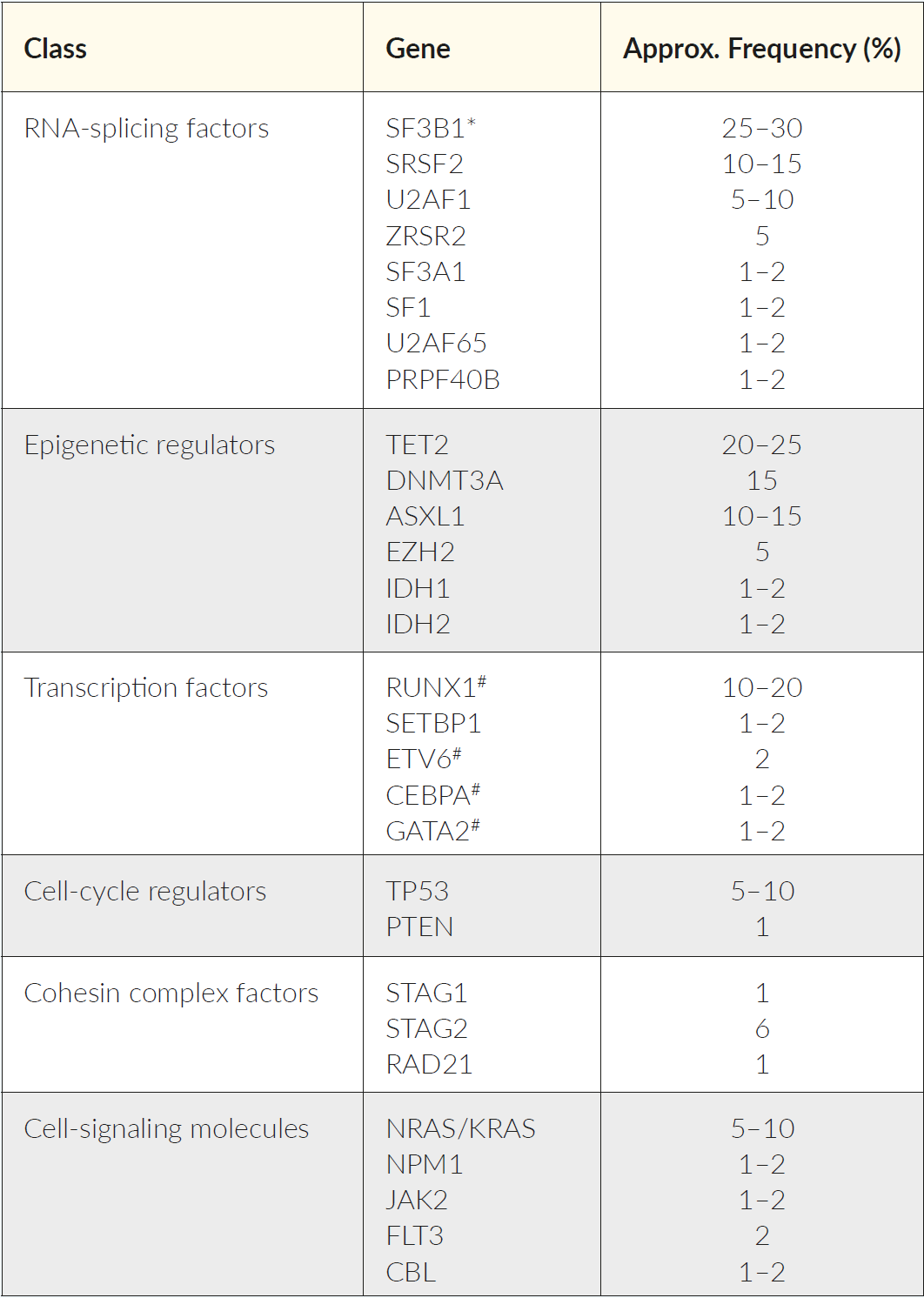
Despite the advances of high throughput sequencing, that allowed to elucidate the genetic background of MDS in the last years, the precise molecular mechanisms remain unclear.8 The hallmark of heterogeneous clonal diseases is the sequential accumulation of genetic lesions in HSPCs.9 Initially, only chromosomal aberrations could be detected by conventional cytogenetics. The advent of next-generation sequencing (NGS) allows to identify recurrent somatic leukemia-associated driver mutations (SLADMs) in an array of genes involved in RNA-splicing, epigenetic regulation, transcription, cell-cycle regulation, cohesin complex, and cell-signaling (Table 1).10–14

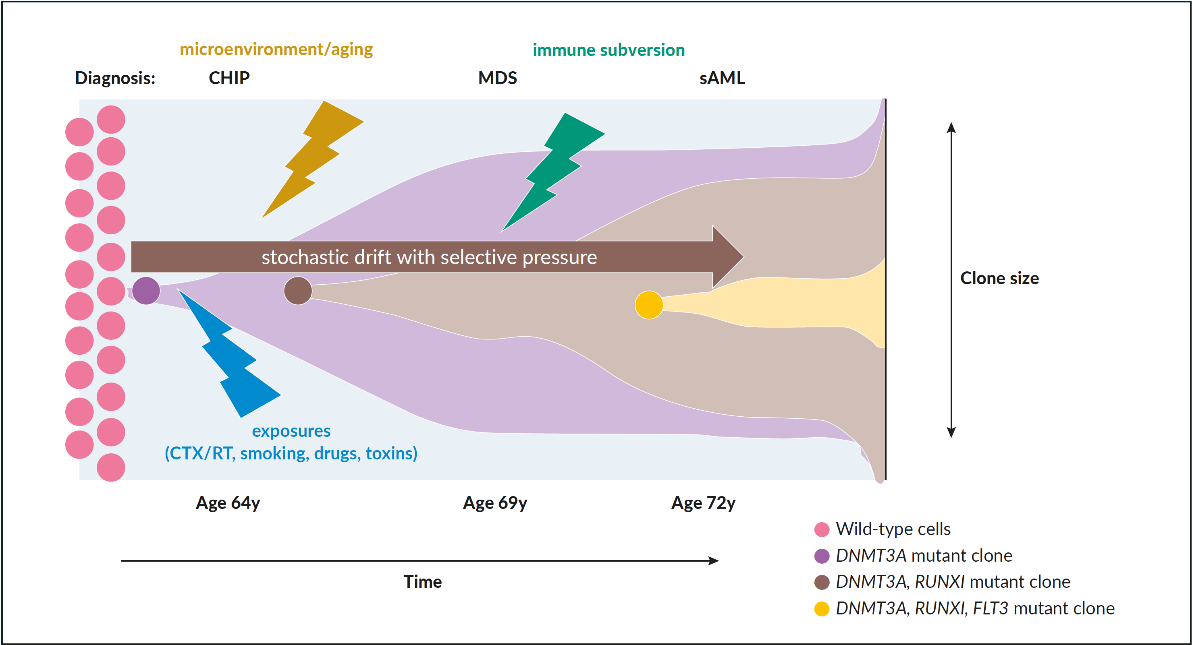
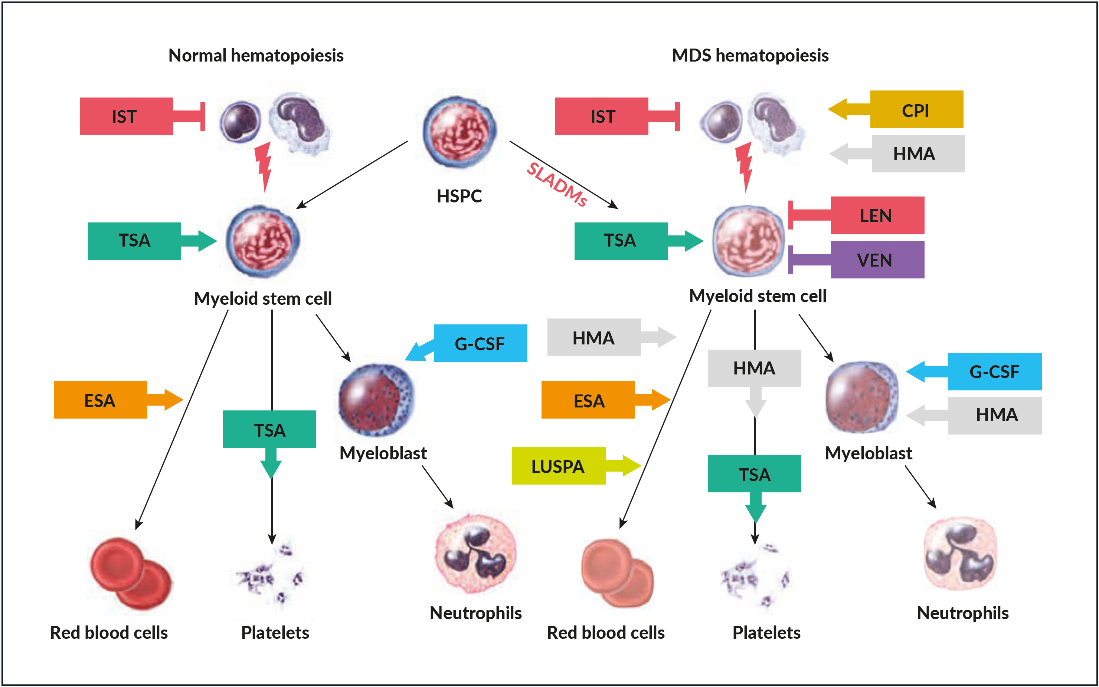
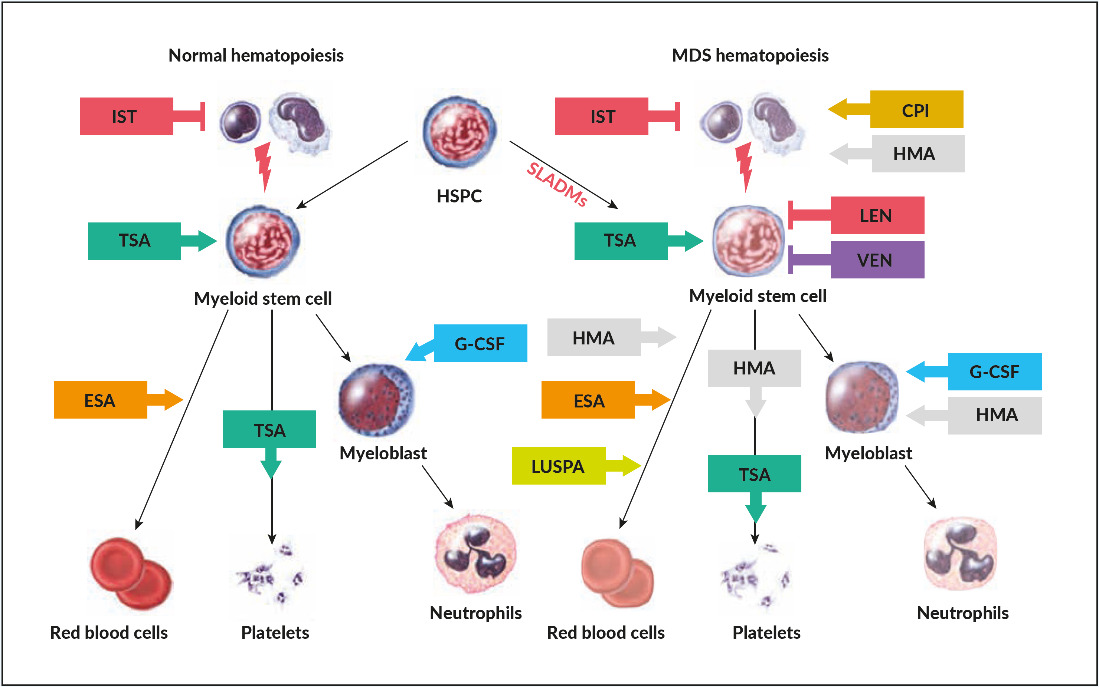
As in many other malignancies, evolution from clonal hematopoiesis, MDS, and secondary AML is caused by a stochastic accumulation of genetic hits in HSPCs with a selective pressure imposed by intrinsic growth advantage, extrinsic factors (radiation, chemotherapy, smoking, drug or benzol exposition etc.) and loss of immunogenic tumor surveillance (Figure 1).15 This process seems to be coupled to coexisting intrinsic deficiencies in repairing DNA lesions, reduced immunogenicity as well as unfavorable micro-environmental factors that are frequently associated with aging.16 More recent investigations have highlighted the fact that clonal hematopoiesis perpetuates a vicious circle of a pro-inflammatory microenvironment caused by NLRP3-activation of the inflammasome, increased production of IL1 beta, and propagation of clonal HSPCs.17 This inflammatory background leads to ineffective hematopoiesis with increased cell death and cytopenias in low-risk disease, which is linked to pyroptosis, a mechanism of cell-death caused by hyper-inflammation.17 Age-related clonal hematopoiesis seems to be associated with increased cardiovascular morbidity including premature atherosclerosis, unfavorable cardiac remodeling and potentially other chronic inflammatory-degenerative disorders of aging.18 The switch from an activated to exhausted immunological tumor surveillance, described as immunesubversion, promotes further clonal expansion. This is a well-known but poorly understood phenomenon observed in cancer biology, which opens the field for novel therapeutic interventions in controlling the progression of the disease.19

DIAGNOSTIC APPROACH
A patient history on potential exposure to genotoxic agents (i.e. chemotherapy, radiation, benzol, pesticides, insecticides, chemical waste) should be obtained in every suspected MDS patient as well as a detailed family history for signs of germline predisposition (malignancies, bleeding, immune dysregulation or specific organ dysfunctions in first and second-degree family members), especially in younger individuals. According to the European Leukemia Net (ELN-2013) recommendations, laboratory analyses are classified as “mandatory” (peripheral blood smear, bone marrow aspirate/biopsy, cytogenetic analysis), “recommended” (fluorescence in-situ hybridization (FISH), flow cytometry immunophenotyping and “suggested” for specific circumstances (single-nucleotide-polymorphism-array, mutational analysis).10
WHO classification and minimal diagnostic criteria for MDS
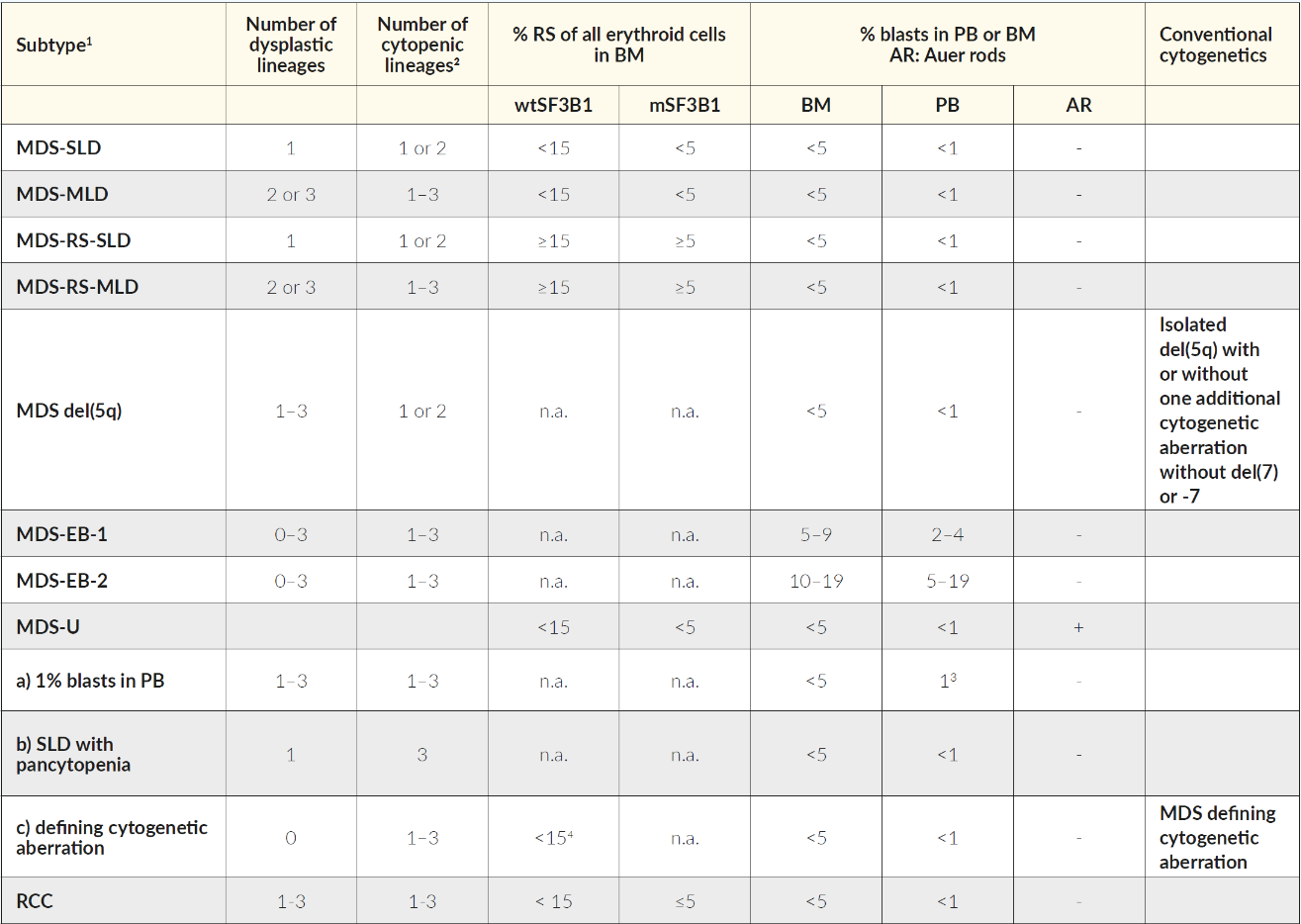
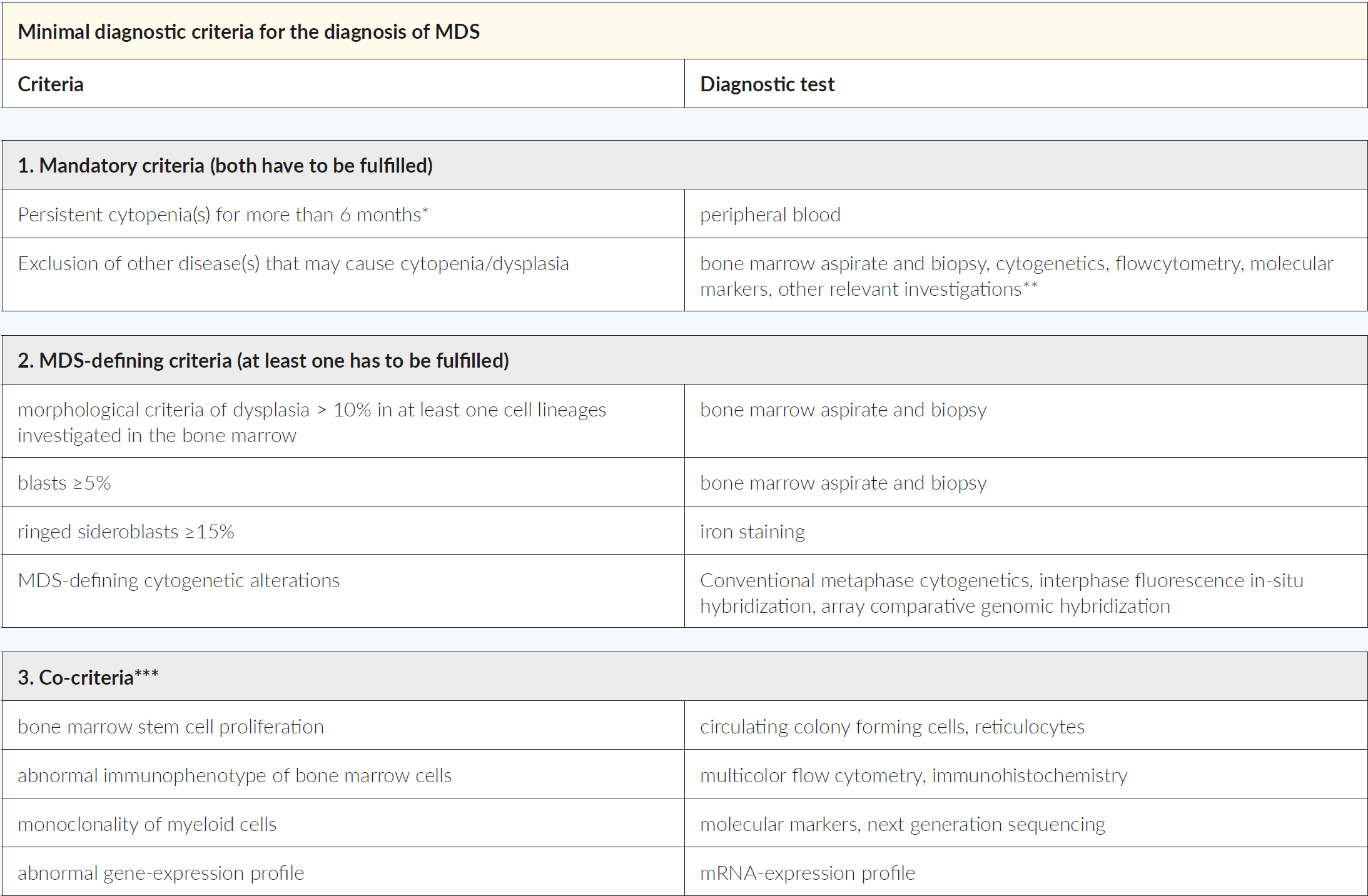
MDS are classified according to the 2016 revision of WHO (Table 2).20 The previously used term of refractory anemia is obsolete because MDS patients may present with cytopenias beyond anemia. Classification of MDS considers the number of lineages affected by cytopenia and dysplasia, the presence of ringed sideroblasts (RS) or mutations in SF3B1 (which is associated with RS), the number of blasts (peripheral blood or bone marrow) and the presence of specific or MDS-defining cytogenetic alterations (i.e. MDS with del 5q). In some cases, the complete hematological work-out of persistent cytopenia (≥6 months), after exclusion of other diseases, may fail to fulfill the MDS-defining criteria (dysplasia >10%, excess of blasts ≥5%, ringed sideroblasts ≥15% or MDS-defining cytogenetic alterations). In such circumstances, an international working group of MDS experts has defined minimal diagnostic criteria that would define conditions with high suspicion of myeloid neoplasm or MDS. For this, co-criteria such as bone marrow stem cell proliferation, abnormal immunophenotype, monoclonality of myeloid cells or abnormal gene-expression profile need to be fulfilled (Table 3).21 If co-criteria are not met, they suggest to classify such conditions as idiopathic cytopenia or dysplasia of undetermined significance (ICUS/IDUS).22


Clonal hematopoiesis of indeterminate potential (CHIP) and Clonal Cytopenia with Unknown Significance (CCUS)
Unexplained anemia is present in about 10−15% of patients over the age of 65 years. Almost 30 years ago, landmark studies identified age-associated skewing in stem cells using X-chromosome inactivation.23,24 More recent studies using NGS identified SLADMs in up to 20−40% of elderly individuals >80 years with normal blood values. These studies underlined the fact that clonal hematopoiesis is an age-related phenomenon and affected individuals showed an increased risk of transformation in overt hematological malignancies.25–27 The condition was subsequently termed Clonal Hematopoiesis with Indeterminate Potential (CHIP) in individuals with normal peripheral blood values and Clonal Cytopenia of Unknown Significance (CCUS) in individuals with cytopenia.28 NGS has substantially improved our diagnostic sensitivity in identifying clonality in patients with unexplained cytopenia. The evolution into overt hematological malignancies depends on the affected genes (high-risk vs lower-risk mutations), clonal burden (>10% variant allele frequency [VAF]), and the number of mutations.29 NGS allows identifying patients at early pre-MDS conditions and the distinction between innocuous age-related changes and initial stages of myeloid malignancies is increasingly blurred. In uncertain circumstances, observation for 3−6 months with repetition of bone-marrow analysis is indicated when clinical or laboratory signs of progression are present.10
GENERAL ASPECTS OF MDS PATIENT MANAGEMENT
The heterogeneity of disease presentation and multimorbidity of elderly MDS patients represents the major challenges to the disease management. The course of the disease varies from chronic asymptomatic or minimal symptomatic cytopenia to rapid progression towards secondary AML. One-third of MDS patients succumb to leukemia, whereas two-thirds die of complications related to bone marrow failure. These include symptomatic anemia, neutropenia, or thrombocytopenia, causing cardiovascular events, infections, and bleeding, respectively.30 Therefore, risk stratification for disease- and patient-based factors is mandatory in each MDS patient. Lower-risk MDS patients have a median survival of 3 to 8 years and die predominantly of non-leukemic causes related to the impact of cytopenia on relevant comorbidities. Thus, treating cytopenia and comorbidities aims to improve quality of life (QoL) and may reduce the risk of progression, which should be the focus for these patients.31,32 Higher-risk MDS patients have a median survival of 1 to 3 years and die predominantly of progression to AML. Treatment should aim to delay progression to AML and improve overall survival.33,34 Guideline-based indicators (GBI) are measurable elements for quality of care and have been recently developed by our Swiss MDS Study Group in collaboration with international experts.35 These GBIs allow the structured and systematic assessment and comparison of quality of care of adult MDS patients in different health care environments.
Disease-based risk stratification
Scoring systems are available to estimate the risk for progression to AML and overall survival. These comprise the International Prognostic Scoring System (IPSS), the revised IPSS (IPSS-R) and the WHO Prognostic Scoring System (WPSS).23,36,37 IPSS and IPSS-Rconsider the number of blasts in the bone marrow, the risk conferred by cytogenetic alterations, and the numbers and severity of the main three cell lineages affected by cytopenia. IPSS was the first scoring system used for clinical trials but has been replaced by the IPSS-R, which is more precise for risk-stratification. Lower-risk MDS is generally characterized by less than 5% of blasts in the bone marrow, no circulating blasts in the peripheral blood, isolated anemia, transfusion independence, and normal karyotype or favorable cytogenetic aberrations. In contrast, blasts more than 5% in the bone marrow, cytopenia of multiple lineages, poor-risk or complex cytogenetic aberrations and transfusion dependency characterize higher-risk MDS.
Patient-based risk stratification
Patient-based risk stratification includes age, comorbidities, frailty and performance status and is important to balance treatment efficacy against tolerability.10 The performance status is assessed by the Karnofsky and Eastern Cooperative Oncology Group (ECOG) scores. As these are influenced by age, a better score for comorbidity is the Sorror Hematopoietic Stem Cell Transplant Comorbidity Index (HCT-CI), which is an adapted version of Charlson Comorbidity Index for patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT).38,39 A simplified version of the HCT-CI is the MDS Comorbidity Index (MDS-CI), which considers only cardiac, hepatic, pulmonary and renal comorbidities as well as prior treatment for solid tumors.40 Patient-based risk factors independently impact on overall survival among MDS patients.41 Furthermore, the evaluation for frailty, functionality, and quality of life in the daily care of geriatric patients would be generally desirable, but the current instruments are not sufficiently standardized, are time-consuming, and their impact on management remains generally unclear.35
THERAPEUTIC APPROACH
Experienced physician should assess MDS patient with symptomatic cytopenia. This is important due to the complexity of this disease, interfering comorbidities, and the selection of patients who might be eligible for the only curative approach of allo-HSCT. Many therapeutic questions remain still insufficiently investigated, and clinical trials are needed.42 For this, symptomatic patients should be referred to MDS centers, included in prospective registries, and offered participation to clinical trials, whenever possible.
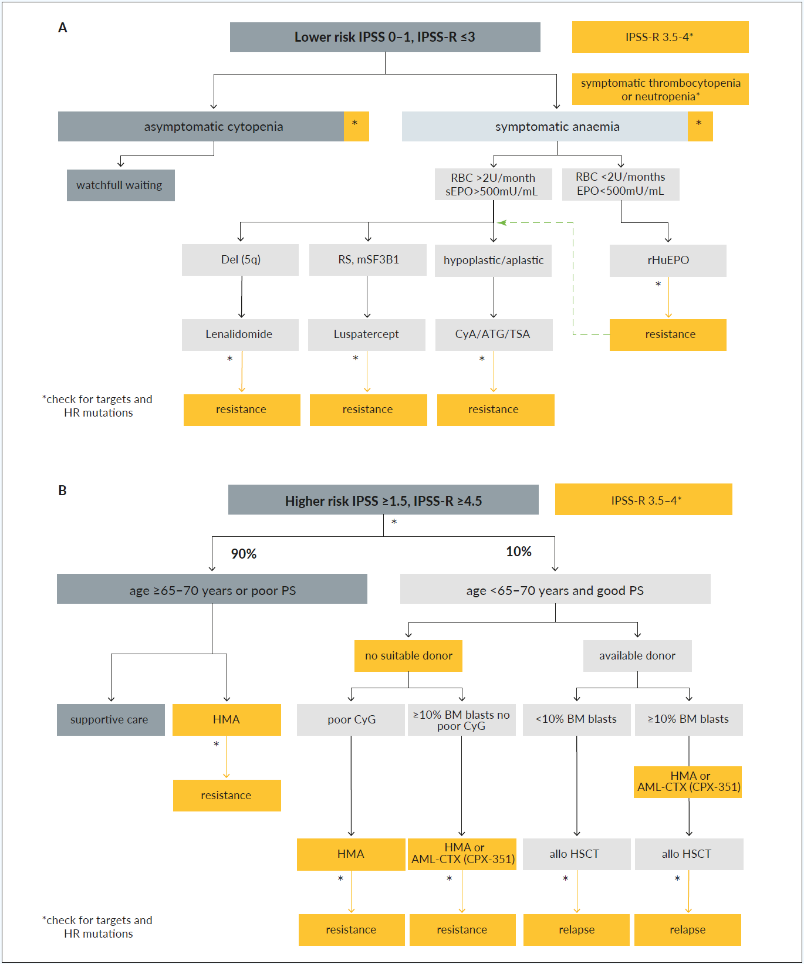
LOWER-RISK MDS PATIENTS (FIGURE 2A AND FIGURE 3)
Watchful-waiting
Patients with lower-risk and asymptomatic cytopenia should not be treated but followed with a regular watch and wait.10 As an example, the life expectancy of asymptomatic patients >70 years of age with MDS-SLD or MDS with del(5q) does not substantially differ from an age-matched population.37 With NGS, lower-risk patients (as for IPSS or IPSS-R) are increasingly identified with ≥3 SLADMs or single mutations in genes with a high risk of AML transformation (HR mutations). These include TP53, RUNX1, ASXL1, ETV6, EZH2, SRSF2, U2AF1, RAS-pathway, and JAK2 at VAF ≥2%.12,43,44 The therapeutic approach to these patients remains not properly addressed in current guidelines or recommendations and require further investigations, as they could potentially profit from interventions that abrogate clonal evolution.
Supportive treatment
Supportive treatment is the mainstay of MDS management and consists of transfusions, iron chelation, growth factors, and infection prophylaxis, which should be complemented by recognition and treatment of nutritional, functional, and psychosocial deficiencies. Properly designed clinical trials in this field are lacking, and the individual physician’s approaches, as a consequence, quite heterogeneous.42

Transfusions
Transfusions of red blood cells (RBC) have been shown to improve quality of life and thrombocytes concentrate (TC) bleeding events. However, there is no established triggers for RBC or TC transfusions in MDS patients. The current consensus is to define individual thresholds for each patient and to apply a symptom-oriented policy. Generally, this means that most patients will receive RBC transfusions at hemoglobin levels <80 g/l or higher in patients with cardiovascular or pulmonary comorbidities. The management of TC transfusions is generally extrapolated from data generated in patients with chemotherapy-induced thrombocytopenia, which might not be fully appropriate for chronic thrombocytopenia.45 Generally, prophylactic TC transfusion will be applied, if thrombocytes drop <10 G/L or <20 G/L with additional risk factors for bleeding, such as fever, mucositis or strictly required anticoagulation. For patients requiring prophylactic anticoagulation, we generally aim for platelet counts >20 G/L and for those with therapeutic anticoagulation >50 G/L. These are empirical cut-off values for an adequate hemostatic function. However, physicians need to be aware that some MDS patients may have cryptic thrombocyte dysfunctions and could be at risk for bleeding events at higher platelet counts.46 RBC transfusions are associated with complications, mainly alloimmunization and iron overload.47 Guidelines recommend irradiation of RBCs in patients that are potential candidates for allo-HSCT, in order to reduce the risk of HLA alloimmunization and transfusion-associated graft versus host disease.48 Controlled trials supporting this approach are lacking. All TCs are generally irradiated for pathogen inactivation and do not need additional measures. However, HLA and HPA alloimmunizations remain of concern for transplant candidates and patients with TC refractoriness.49
Iron chelation
The actual consensus extrapolates the recommendation from patients with thalassemia and suggests starting iron chelation therapy (ICT) after transfusion of more than 20 RBCs and ferritin levels of >1,000 mg/l. Moreover, transfusion-dependent patients eligible for allo-HSCT and a life expectancy of at least one year should receive ICT according to expert consensus.10,50,51 A recently performed phase 3 study in MDS patients with transfusion-dependant iron overload undergoing ICT (TELESTO) unfortunately failed to recruit sufficient participants. This substantially limited the initially planned power and left the study only with the possibility to identify a reduction in EFS (mainly cardiovascular events).52 Therefore, the benefit of ICT remains controversial, and approaches in patients with secondary iron overload are heterogeneous.35,53,54 Because of tolerability and practicability, the currently preferred ICT is deferasirox for the treatment of iron overload in MDS. Kidney insufficiency may be limiting for desferasirox in many elderly patients.
Growth factors and maturating agents
Treatment with erythropoietin stimulating agents (ESA) is recommended for lower-risk MDS patients with symptomatic anemia. Best responses are achieved in those patients with hemoglobin levels of <100 g/L, serum erythropoietin of <500 U/L and transfusions of <2 RBC units/month.10,55,56 All currently used ESAs (recombinant human erythropoietin alpha and beta or darbepoetin alpha) provide similar erythroid responses (ESA-naïve, 45−73%; previous ESA exposure, 25−75%) and duration of response (1−2 years) in equivalent doses.57 The initial dose is usually at 30,000 U/week sc (approximately 150 µg darbepoetin alpha/week or 300 µg/2 weeks), which is markedly higher than in patients with renal insufficiency. The dose should be doubled after 8 weeks in case of lack of response. The ELN 2013 guidelines suggest adding granulocytic-colony stimulating factor (G-CSF at 3x 300−480 µg/week sc) in patients with insufficient erythroid response to ESA after 8−12 weeks.10,58,59 However, convincing data for the additional efficacy of G-CSF in patients treated with full-dose ESA (60,000 U equivalents/week) are lacking.60 Patients refractory to ESA have a worse prognosis and may be candidates for clinical trials, hypomethylating agents (HMA) or allo-HSCT.61
Luspatercept is a recombinant protein with a modified extracellular domain of the activin receptor IIb fused to the human Fc domain of IgG1. It acts as a ligand trap that neutralizes TGF-ꞵ superfamily molecules. The substance interferes with aberrant Smad2/3 signaling involved in ineffective erythropoiesis and enhances late‐stage erythroid maturation by reducing the negative effect of Smad 2/3. This novel class of substances are called erythroid maturating agents (EMA). In an initial phase II (PACE) and the subsequent phase III, randomized, placebo-controlled trial (MEDALIST), about 1/3 of ESA refractory or ineligible patient with MDS with RS achieved transfusion independency, with a median duration of cumulative transfusion independence of 79.9 months, while 2/3 of patients experienced an erythroid improvement.62,63
Thrombopoietin-stimulating agents (TSA) (i.e. romiplostim, eltrombopag) have shown to raise the platelet counts and reduce bleeding events in phase II and III randomized trials in lower and higher-risk MDS patients.64–66 There was no impact on the rate of progression to AML or on overall survival, but the time to follow-up seemed not to be sufficient to drive sound conclusions.67 As such, there are still controversies about which patients may profit from TSA and, due to some observation of increase in blast counts, there is reluctance in using these compounds in higher-risk MDS patients.35
Regarding neutropenia, sufficient evidence and consensus regarding the prophylactic use of G-CSF or antibiotics in patients with severe neutropenia (ANC <0.5 G/L) are lacking. In analogy to thrombocytes, neutrophilic dysfunction can occur with normal absolute counts, leading to susceptibility to infections. Using G-CSF or antibiotics for primary prophylaxis in severe neutropenia is generally not recommended, whereas for secondary prophylaxis the decision should be individualized. Primary antimitotic (fluconazole, posaconazole), pneumocystis jirovecii (cotrimoxazole), and antiviral (valacyclovir) prophylaxis is frequently used in patients with agranulocytosis, especially for those patients undergoing intensive chemotherapy or combined cyclosporine A (CyA) and anti-thymocyte globulin (ATG) treatment.68 Finally, most MDS patients receive vaccination against influenza and pneumococcus. The immunologic response might be variable but seems not to be affected by treatment with HMA.69
Disease-modifying treatment
Immunomodulatory drugs (IMIDs)
Thalidomide was found to reduce transfusion need in some MDS patients.70 Lenalidomide (Revlimid®), a derivate of thalidomide, lacks the undesired neurologic side effects. It exhibits anti-cytokine, anti-angiogenetic effects and is synthetically lethal for casein-kinase 1 haploinsufficient cells in del(5q).71 In a phase III trial in transfusion-dependent, lower-risk MDS patients with del(5q), lenalidomide showed sustained transfusion independence in two thirds and cytogenetic responses in half, with a median duration of transfusion independence of one and a half years.72 Response should be assessed after 4 months of treatment and stopped if treatment targets are not achieved. Mutations in TP53 are present in about 20% of MDS del(5q) patients at diagnosis and are associated with lower response and higher risk for progression.73,74 In these patients, the choice remains open between lenalidomide and other therapies, such as HMAs and allo-HSCT.10 As of adverse effects, severe neutropenia and thrombopenia can occur during the first weeks of treatment and require close monitoring of blood values. Currently, lenalidomide is not licensed for the treatment of patients with non-transfusion dependent del(5q) MDS ,transfusions dependent patients with non-del(5q) MDS or in the ESA-refractory setting. The combination of lenalidomide and ESA seem to provide some additional erythroid responses in the ESA refractory setting but further clinical trials are warranted.75
Immunosuppressive treatment
Patients with hypoplastic MDS represent 10% of all MDS cases and can be distinguished from aplastic anemia using histopathological, cytogenetic and molecular criteria.76 Treatment with ATG combined with CyA can lead to long-lasting responses in 16−67% of cases and depends on careful preselection.77 Predictors of response are hypoplastic BM, younger age (<60 years), lower-risk score, normal karyotype or trisomy 8, presence of HLA-DR15, absence of RS, female gender, and short duration of transfusion requirement.77 The current ELN-guidelines recommend ATG with 6 months CyA in younger MDS without a strict recommendation regarding the duration of therapy.10 Relapses have been observed upon the termination of CyA treatment and, therefore, therapy should be continued as long as effective and tolerated.78 The combination with eltrombopag has shown to increase response rates in aplastic anemia but is not approved for hypoplastic MDS.79
HIGHER-RISK MDS PATIENTS (FIGURE 2B AND FIGURE 3)
Hypomethylating agents
HMAs comprise the pyrimidine nucleoside analogs 5-azacytidine (AZA) and 5-aza-2’deoxycytidine/decitabine (DEC); DEC is approved in the US but not in Europe for treatment of MDS.80,81 AZA is well-tolerated and has demonstrated significant higher partial and complete responses as well as a survival benefit (median OS 24.5 vs 15.0 months) in a phase III trial compared to best supportive care (hydroxyurea and low dose AraC).80 Responses were independent of age, marrow blast percentage and karyotype. As responses are often observed after a median treatment duration of 3 months, it is advised to wait for at least 6 cycles to consider a patient as resistant. HMA remains inferior to more intensive induction chemotherapy followed by allo-HSCT, for which, however, only younger and fit patients are eligible. MDS with complex karyotype, either in elderly or younger patients, without a suitable stem-cell donor, should preferentially be treated with HMA, due to the lower rates of complete remission and higher toxicity imparted by intensive chemotherapy in this setting.82 Markers for response prediction to HMA have not been identified, however, lower response rates have been described for patients with poor performance status (ECOG >2), high transfusion dependency (>4 RBCs over 8 weeks), higher numbers of bone marrow blasts (>15%), circulating blasts, higher cytogenetic risk scores and TP53 mutation. In contrast, mutations in epigenetic regulators such as TET2, EZH2 and DNMT3A seem to be associated with better responses.83–86 As HMA does not modify significantly the clonal composition of the disease, treatment should be offered as long as tolerated in absence of signs of progression. There is currently no established treatment after failure to HMA and these patients should be offered clinical trials.
Induction treatment
Cytoreductive induction treatment with AML-based chemotherapy before allo-HSCT is the mainstay for young and fit higher-risk MDS patients with ≥10% bone marrow blasts.87 Alternative induction treatments are fixed liposomal combinations of danorubicine/cytarabine (CPX-351) or HMA in elderly patients that are deemed to be eligible for allo-HSCT but at increased risk of toxicity for standard induction chemotherapy. However, properly designed clinical trials are still lacking for this specific question. Good prognostic factors for allo-HSCT are younger age, good performance status and favorable cytogenetics [54]. For higher-risk MDS patient with <10% bone marrow blast, it remains controversial whether HMA induction is required or it is better to proceed directly to allo-HSCT.87 For patients without a suitable donor and ≥10% bone marrow blasts one course of induction chemotherapy may be recommended followed by HMA maintenance.10 Patients with poor-risk cytogenetics or mutations in TP53 should be preferentially treated with HMA, as toxicity predominates the limited responses to standard chemotherapy.82,88 These patients should be offered induction treatments within clinical trials.
Allogeneic HSCT
Allo-HSCT remains the only curative option for the minority of fit, higher-risk MDS patients up to 65−70 years of age.87 The oldest patient that has ever been transplanted in Switzerland was 75 years old. As non-relapse mortality is dependent on comorbidities and occurs in approximately a third of these patients, the Hematopoietic Cell Transplant Comorbidity Index (HCT-CI) is relevant to estimate non-relapse mortality and has to be used as patient-based factor for selection of appropriate candidates.39 Maximal benefit of allo-HSCT is associated with transplantation in patients in the higher-risk disease state, whereas lower-risk patients with poor-risk cytogenetic/genetic features, profound cytopenias, and high transfusion burden may also profit from transplantation.89 Age, disease status and molecular gene status are the most important predictive factors for overall survival after allo-HSCT, with TP53, RAS pathway, ASXL1, RUNX1 mutations associated with a higher risk of relapse.90–93 Reduced-intensity conditioning regimens are considered for patients with comorbidities or age >50 years; however, prospective randomized clinical trials have not provided sufficient evidence for the optimal conditioning regimen.94,95 All MDS patients should be offered maintenance therapy and MRD-based consolidation therapy after allo-HSCT, whenever possible in the context of clinical trials.10
FUTURE DIRECTIONS
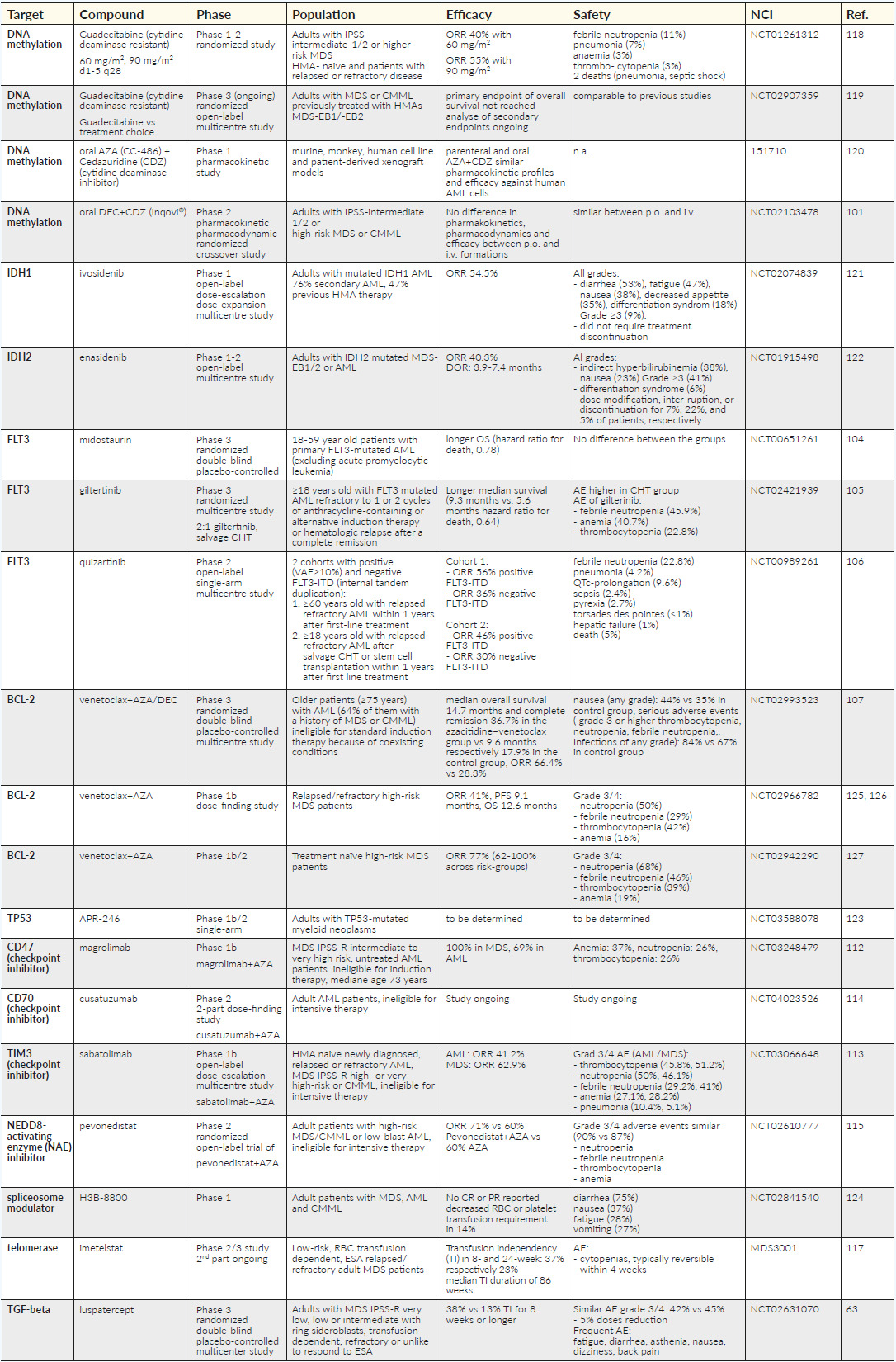
Longer exposure is considered to potentially improve the efficacy of HMAs, as they act in the S-phase of the cell cycle. Therefore, there are strategies to prolong half-time or provide oral applications to achieve this aim. Cytidine deaminase resistant HMAs have been developed (guadecitabine)96–98 as well as oral formulations of AZA (CC‐486)99,100 or fixed dose combinations of oral decitabine with the cytidine deaminase inhibitor, cedazuridine.101 CC-486 has been approved by the FDA for maintenance treatment in AML, while cedazuridine/decitabine received approval as first line treatment in MDS; these agents are further investigated in the MDS setting.

The discovery of SLADMs has opened a completely new era for risk stratification and patient selection for target therapies in AML, which is increasingly translated also into clinical trials in higher-risk MDS setting. IDH1 and IDH2 mutations are rare in MDS and the inhibitors, ivosidenib and enasidenib, respectively, have shown encouraging results in AML patients.102,103 Other target therapies that may be taken over from AML treatment include the FLT-3 inhibitors (midostaurin, gilteritinib, quizartinib)104–106 and the BCL2 inhibitor, venetoclax, in combination with azacitidine or decitabine.107 Venetoclax with HMA is currently investigated in the first line and HMA relapsed/refractory MDS setting.108 A very promising compound is APR-246, a reconfirming agent of mutated TP53, which has shown unanticipated responses in TP53 mutated AML and MDS patients.109–111 Other drugs that are tested in combination with HMA are checkpoint inhibitors (CD47-Ab: magrolimab, CD70-Ab: cusatuzumab, TIM3-Ab: sabatolimab) and the NEDD8-activating enzyme (NAE) inhibitor,pevonidostat.112–115 Other compounds that are under investigation in higher-risk MDS include oral spliceosome modulators (H3B-8800)116 and in lower-risk,ESA refractory MDS patients, the telomerase inhibitor imetelstat as well as ESA combinations with luspatercept, which some of these trials that are about to be launched also in Switzerland.117
CONCLUSIONS
The recent drug developments are encouraging for MDS patients. However, none of those will provide the Holy Grail that rejuvenates the stem cell compartment with abrogation of clonal evolution. Therefore, further investigation on the mechanisms of disease and clonal evolution remain relevant to substantially advance our understanding of MDS. Improvement of treatment allocation, based on efficacy and tolerability, is a cornerstone of MDS management. The inclusion of MDS patients in biobanks, as well as prospective national and international registries, is highly encouraged to advance translational research, improve health care provided and identify patients who could profit from inclusion into the increasing numbers of clinical trials in MDS.
TAKE-HOME-MESSAGES
-
Myelodysplastic syndromes (MDS) incident cases are rising with a relevant impact on health care resources.
-
Experienced physicians should assess MDS patients to provide the most appropriate management plan.
-
NGS allows identifying early stages of clonal hematopoiesis and monitoring of clonal evolution.
-
Allo-HSCT remains the only curative treatment option for the minority of eligible MDS patients.
-
New treatment options are on the horizon, but abrogation of clonal evolution remains out of reach.
CONFLICT OF INTEREST
The authors declare no conflict of interest for this review article. Potentially perceived conflicts of interests according to the definitions and terms of the International Committee of Medical Journal Editors are IC: none; NB: Amgen: financial support for travel; Astellas: research funding to institution; Celgene/BMS: advisory board, consultancy, financial support for travel, research funding to institution; Janssen: financial support for travel; Novartis: financial support for travel, research funding to institution; Roche: financial support for travel, research funding to institution; Sandoz: advisory board, research funding to institution; Servier: research funding to institution; Takeda: advisory board.
Author Contributions
All authors contributed to and approved the final manuscript.