




INTRODUCTION
Healthcare systems have focused for the last 10 years on addressing patient care, leaving the concept of a one‐drug‐fits‐all model by identifying aspects inherent to patients and their diseases that might allow diagnosis and management optimization by offering customized care to each case. This healthcare model called precision medicine or personalized medicine is considered as particularly relevant for those diseases in which a marker can have a prognostic meaning or can represent a target for specific treatment.1 Hence, this model represents a current modern approach in the management of malignant diseases in general and thanks to the enormous contribution of molecular and genomic in hematological diseases in particular.2
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders characterized by activation of the physiologic signal-transduction pathways responsible for cell proliferation of one or more of the hematopoietic lineages. The diverse clinical features that occur in each MPN entity with variability in risks of disease complications and progression to secondary leukemia are challenging for physicians taking care of these patients. In MPNs, consideration of individual variability is imperative in diagnostics and management of the disease. The clinical presentation, the peripheral blood values and clinical chemistry parameters, the bone marrow’s histological features as well as the molecular/genomic profile are all aspects that might contribute to a more precise way to diagnose and treat these patients.3 This review attempts to summarize all those aspects that can contribute to the most detailed diagnosis and therapy approach in patients with classic BCR-ABL1 negative MPNs, emphasizing the relevance of interpretation of each step in the concept of personalized medicine.
BLOOD COUNTS IN MPN
In MPNs, a comprehensive evaluation of the peripheral blood counts reveals characteristic changes that can guide the diagnostic process from the very beginning. The presence of polyglobulia alone or accompanied by leukocytosis and thrombocytosis suggests the existence of an underlying polycythemia vera (PV). Isolated thrombocytosis is the classic presentation of essential thrombocythemia (ET) or a pre-fibrotic form of myelofibrosis (MF). MF in its initial form can present any type of cytosis, advanced forms in opposite are characterized by cytopenias. None of these peripheral blood changes are, however, specific for a distinct MPN. At presentation, cytosis in the context of an MPN should be differentiated from cytosis occurring due to reactive causes, which are significantly more frequent. In cases of presentation with cytopenia, the characteristic changes in the peripheral blood smears may help to guide towards the correct diagnosis, in these cases the main differential diagnosis is myelodysplastic syndrome (MDS).
The red blood cells (RBC) on their own can be informative. Michiels et al. (1997) could prove that an RBC count within the normal range (<5.8×1012/L in males and <5.6×1012/L in females) may distinguish JAK2V617F positive ET from prodromal PV and overt PV.4
It has been demonstrated that changes in red cell distribution width (RDW) in patients with PV tend to be increased than in the normal controls when RBC were high.5 The presence of microcytosis in patients with high hemoglobin (Hb) levels could also be suggestive of PV associated with iron deficiency. Virtually all PV patients might have iron deficiency during the course of the disease. Iron metabolism in PV may be altered due to aberrant erythropoiesis, an inflammatory milieu, decreased systemic iron concentration, and potentially altered hypoxia responsiveness directly influencing iron absorption in the small intestine.6
Sandes et al. (2017) retrospectively analyzed 248,839 outpatients without known hematological disorders7 and were able to prove that the isolated use of the World Health Organization (WHO) proposed Hb/hematocrit (Hct) levels as a definer of polycythemia may lead to a substantial increase in unnecessary diagnostic tests.8 In cases with borderline levels of Hb, the diagnostic workup of PV should only be initiated in the presence of clinical and/or laboratory features associated with MPNs.
In MPNs, the evaluation of blood smears may reveal leukocytosis, eosinophilia, basophilia, or the presence of immature myeloid forms. Likewise, quantitative or qualitative abnormalities of platelets are frequently observed, showing anisocytosis with large platelets and platelet degranulation. A leucoerythroblastic picture (i.e., teardrop-shaped RBC, circulating nucleated RBCs and/or immature myeloid cells) while not being pathognomonic, may suggest an advanced form of myelofibrosis.
SYMPTOMS IN MPN PATIENTS
The spectrum of MPNs-related symptoms is wide. These include constitutional symptoms (fever, night sweats, and weight loss), symptoms related to spleen enlargement (abdominal discomfort or pain and early satiety), symptoms related to microvascular disturbances (vertigo, lightheadedness, dizziness, insomnia, sexual dysfunction, numbness, tingling, headaches, and concentration problems), as well as fatigue, cough, bone pain, inactivity, and pruritus. Symptom heterogeneity may exist within each MPN subtype, sometimes independent of disease features or prognosis.9
Symptomatology of the patients can be assessed through the MPN Symptom Assessment Form (MPN-SAF or MPN-10) questionnaire, a 10-item survey/symptom score, which has proved effective in capturing the presence and intensity of MPN-related symptoms.10 Each symptom is rated on a potential score of 0/absent to 10/worst imaginable. These ten core items include worst fatigue, early satiety, abdominal discomfort, concentration problems, inactivity, night sweats, itching, bone pain, fever, and weight loss. Although these symptoms are not taken into consideration in the WHO classification, using the MPN-10 score at each visit is an excellent way of objectifying the clinical follow-up in patients with MPNs. Thus, the presence of symptoms interfering with patient’s quality of life should suggest the need to initiate or modify treatment.
Disease‐related PV symptoms, such as microvascular disturbance, pruritus (which may be excruciating), migraine‐type headache, and fatigue, can significantly impact the quality of life.11 However, patients may as well be asymptomatic at presentation. Constitutional symptoms in early PV are unusual, except for fatigue, which may have many causalities, including iron deficiency, thyroid or cardiovascular disease, sleep apnea, depression and pulmonary hypertension, and side effects related to the therapy used to treat the disease.12 Geyer et al. showed that patients with problematic PV (specifically hydroxyurea treatment failure, splenomegaly, or a persisting need for phlebotomies) have worse symptom burden compared with patients without these features.13
Most patients with ET are asymptomatic at diagnosis, and detection of thrombocytosis is typically incidental. Others present with disease-related vasomotor symptoms affecting microcirculation (e.g., headache, dizziness, visual changes, acral paresthesia) or thromboembolic or bleeding complications (e.g., bleeding first-trimester fetal loss). Unlike in PV, pruritus is very uncommon in ET, occurring in less than 5% of patients. Although ET has the lowest symptom severity, the prevalence of constitutional symptoms reported by patients is relatively high.12 Patients should be advised to modify their lifestyle where necessary, including stopping smoking, and should be recommended weight control and treatment of dyslipidemia and arterial hypertension if necessary. MF patients most frequently complain of severe fatigue, symptoms due to an enlarged spleen, as well as weight loss, low-grade fever, bone pain, and night sweats.14,15 Up to 30% of patients are initially asymptomatic. As the disease evolves, all patients become symptomatic due to marrow failure, increasing splenomegaly, and constitutional symptoms. In the advanced phases of MF, extramedullary hematopoiesis in sites other than the spleen and liver may be seen.
MPN ENTITIES
According to the WHO, the combination of clinical, morphological, and molecular genetic features is the most suitable attempt to define MPNs, with the importance of bone marrow morphological features being highlighted. Bone marrow morphology remains the central distinguishing feature in the 2016 WHO classification of MPNs.8
POLYCYTHEMIA VERA
PV is characterized by a molecular signature; all patients harbor the JAK2 mutation, exceptionally in exon 12. Although other MPN entities can as well carry a JAK2 mutation, the characteristic high allele burden can contribute to guide the diagnosis towards PV already at the time of first detection. Recently, another mutation was identified in JAK2, which is characterized by a common 4-amino-acid deletion and variable 1-amino-acid insertion (Leu583-Ala586DelInsSer/Gln/Pro) within the JH2 domain. This novel recurrent mutation, albeit rare, was described in patients with eosinophilia, which fulfilled diagnostic criteria of both PV and chronic eosinophilic leukemia (CEL).16
The main changes in the revised 2016 WHO criteria are the Hb/Hct threshold, the upgrade of bone marrow (BM) biopsy to a major criterion together with the presence of JAK2V617F exon 14, or JAK2 exon 12 mutations. The subnormal serum erythropoietin level remains as a solely minor criterion since endogenous erythroid colony assay is no longer considered as a minor criterion.1 In the new classification, special importance is given to the histological evaluation of the BM in all MPNs, granting it a major criterion attribute for all MPN subtypes, including PV. In PV, the findings on BM examination change as the disease evolves from a prodromal pre-polycythemia phase with borderline to mild erythrocytosis to an overt polycythemic phase with increased red cell mass to a “spent” or post-polycythemic myelofibrosis with cytopenias, ineffective erythropoiesis, fibrosis, extramedullary hematopoiesis, and hypersplenism.17
MASKED PV
MPN patients may present with clinical features suggestive of PV such as abdominal thrombosis (Budd Chiari syndrome), isolated thrombocytosis, leukocytosis, or splenomegaly, but with Hb levels that do not reach the threshold defined by WHO. The lack of clear evidence of polyglobulia, may guide the diagnosis towards other MPNs than PV.
Masked PV (mPV) was initially described as inapparent PV or latent PV.18 Lamy et al. (1997) observed that in certain situations, Hb and Hct levels remain normal even though there is a genuine case of polycythemia. A study conducted by Barbui et al. describes mPV as a possible variant of PV with lower Hb levels at baseline but with a trend to more frequent thrombotic events as well as more rapid progression to myelofibrosis and acute leukemia.19 Distinguishing ET from masked PV based on Hb or Hct values alone is rather impossible, yet this distinction is of utmost importance. Alvarez-Larran et al. (2012) showed that mPV could be identified using red blood cell mass (RCM) measurement.20 Cassinat et al. (2008) showed that a proportion of patients harboring JAK2 mutation had in fact early-stage polycythemia when RCM was systematically measured, these patients were erroneously classified as ET.21 The distinction between PV versus ET is indeed of clinical relevance with therapeutic impact since these patients should be managed as PV and not as ET, giving priority to Hct control, keeping it below 45% at least. This data underscores that in the era of molecular classification, the clinic continues to play a relevant role in the classification of MPNs.18 Other studies contributed to confirm this data, showing that once RCM is estimated, masked PV can turn out to be rather “unmasked” and a true PV.22
PV may also present normal Hb values in some special situations, e.g., portal hypertension secondary to suprahepatic or portal vein thrombosis may present an expanded plasma volume that masks an increased RCM.23
ESSENTIAL THROMBOCYTHEMIA
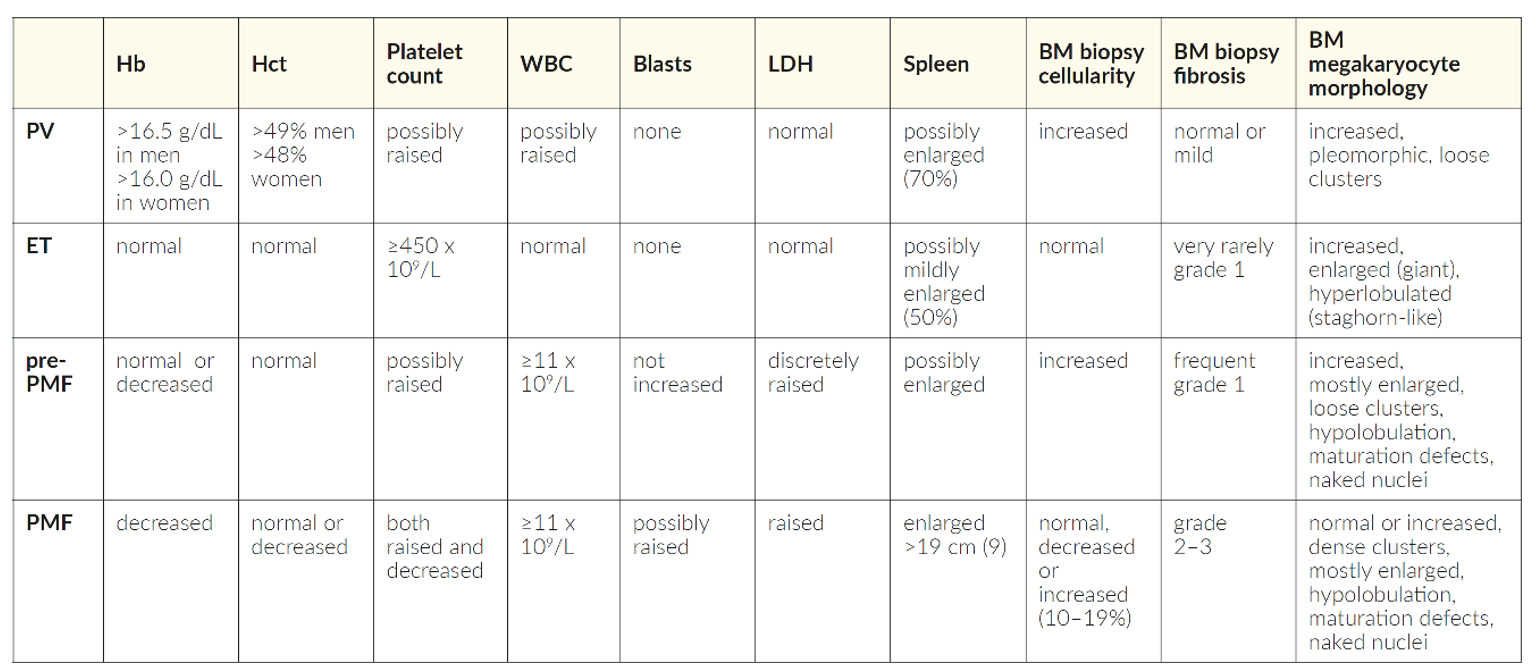
ET is characterized by excessive clonal platelet production with a tendency to thrombosis and hemorrhage. Distinguishing reactive thrombocytosis from thrombocytosis associated with an underlying MPN is a critical step in patient’s care. Since thrombocytosis may be the only abnormal finding, molecular testing should be performed for JAK2, CALR, and MPL. Especially in triple-negative cases, BM evaluation is necessary to identify the correct diagnosis. In ET, BM cellularity, size, and morphology of megakaryocytes contribute significantly to the diagnosis (Table 1). Patients with reactive thrombocytosis show neither spontaneous megakaryocyte nor erythroid colony growth.13
ESSENTIAL THROMBOCYTHEMIA VERSUS PREFIBROTIC/EARLY PRIMARY MYELOFIBROSIS
The distinction between prefibrotic myelofibrosis (pre-PMF) and ET may be difficult since both entities depict considerable overlap. The tendency of megakaryocytes to form clusters, as well as BM reticulin or collagen fibrosis, are both characteristics of pre-PMF but not ET helping, therefore in this differentiation (Table 1). Discrimination of ET from the pre-PMF still rests largely on proper histologic BM evaluation,24 as being highlighted in the revised WHO classification.8 Discrimination of both entities is clinically relevant because pre-PMF has a much higher probability of progressing to myelofibrosis and transforming into secondary acute leukemia compared to ET.25,26 Additionally, a JAK2V617F allele burden >50% may favor a diagnosis of pre-PMF.24
PREFIBROTIC/EARLY PRIMARY MYELOFIBROSIS VERSUS PRIMARY MYELOFIBROSIS (OVERT FIBROTIC STAGE)
The distinction of pre-PMF from overt PMF due to their different survival and risk of disease progression has clinical relevance.27 Guglielmelli et al. analyzed data of 661 PMF patients and clearly showed differences between both entities. Patients with pre-PMF are younger compared with those with overt-PMF.28 Patients with overt-PMF have greater hematologic abnormalities (anemia, leukopenia, thrombocytopenia, higher blast counts), more frequently show clinical symptoms, have larger spleens, and have a higher frequency of molecular high-risk profiles.27 With this information, the clinician will be able to categorize patients more correctly and therefore optimize treatment decisions. This further augments the importance of BM evaluation in all patients newly diagnosed with MPNs.
BIOCHEMISTRY
In ET, signs of hypermetabolism with increased lactate dehydrogenase (LDH) and uric acid levels are infrequent,29,30 while they are typical in PMF.31 Mazzotta et al. (2006) found that the mean of LDH level was moderately increased in patients with PV and ET and markedly increased in PMF when compared with secondary polycythemia.32 This is important since increased LDH in PV and ET is associated with increased bone marrow proliferation and increased risk of progression to myelofibrosis and is also an independent predictor of survival.33,34 Considerable elevation of serum LDH independently predicts shorter overall and leukemia-free survival in PMF as well.35
Chronic inflammation has been associated with MPNs.36 In a study of 244 patients with PV and ET, the major thrombosis rate was significantly increased in patients with increased acute-phase protein high sensitivity C-reactive protein (CRP) levels. The highest levels of this biomarker doubled the risk of thrombosis. Conversely, high pentraxin 3 (PTX-3) levels are associated with lower rates of thrombosis.37 Patients with high CRP levels exhibit an increased risk of progressive disease and 3–4-fold increased mortality rate.38 These easily measurable biomarkers might, therefore, be useful to improve the prognostic classification of MPN patients.
In 3–14% of MPN patients, a monoclonal gammopathy can be identified,39–42 probably coincidentally40 though one study has reported its presence to be associate with an adverse MPN disease course.42
BONE MARROW FOLLOW-UP
Bone marrow morphology remains the cornerstone of MPN diagnosis,8 but is also important during the follow-up under therapy. Successful interferon therapy for myelofibrosis has been associated with a significant reduction of marrow fibrosis, cellularity, megakaryocyte density, and naked nuclei density.43 Similarly, ruxolitinib therapy may reduce the amount of bone marrow fibrosis in a large proportion of patients with MPN-associated MF or may delay its progression.
MPN-ASSOCIATED COMPLICATIONS
BLEEDING
Patients with any type of MPN can develop bleeding complications,44 and bleeding can even be the initial presentation of MPN, conducting to the MPN diagnosis.44,45 The prevalence of bleeding among patients newly diagnosed with MPN varies considerably across studies.46–50 A recent review and meta-analysis involving a total of 13,436 patients with MPN reported that a pooled prevalence of bleeding at a diagnosis of 6.2%.51 Minor or major hemorrhage events may also occur during the course of the disease.44
The underlying pathophysiological involved mechanisms are frequently multifactorial. Since MPNs mainly affects elderly patients, comorbidities and polypharmacy may contribute to bleeding complications.
Within the disease-specific risk factors for bleeding, thrombocytosis is a leading risk factor.52 Bleeding risk may be worsened by the development of an acquired von Willebrand syndrome (aVWS), which may result from increased proteolysis with platelet activation leading to reduced von Willebrand factor activity.53 Some studies suggest consideration of aVWS even in the absence of extreme thrombocytosis.54 Thus, investigation of aVWS should be considered before administration of aspirin. CALR mutation has been associated with higher levels of thrombocytosis. However, in a retrospective study, the mutational status (CALR vs JAK2V617F) did not predict bleeding complications.52 Leukocytosis has been identified as bleeding risk factor mainly in patients with ET and early PMF.55
Treatments used in MPN patients can also trigger bleeding complications. Data evaluating the risk of bleeding of antithrombotic treatment is, however, controversial. Vitamin K antagonists (VKA) with or without aspirin did not increase the risk of bleeding in two large studies,50,56 whereas another study demonstrated a trend towards a higher rate of major bleeding in MPN patients treated with VKA compared to those not anticoagulated.57
The use of low dose aspirin to treat MPNs may be associated with increased bleeding complications. However, low-dose aspirin did not affect the risk of bleeding in PV patients in an European multicenter trial.58
Bleeding related to cytoreductive treatments, either hydroxyurea, interferon, or ruxolitinib, might be due to thrombocytopenia while bleeding under anagrelide therapy is more likely related to altered platelet function.59
In order to prevent bleeding in MPN patients, it might be wise to conduct aVWS investigation before prescribing low dose aspirin when the platelet count is above 1000×109/L and to consider testing for aVWS even with lower thrombocytosis. A cytoreduction to lower the platelet count can be administered in the presence of aVWS. Supportive therapies such as desmopressin and von Willebrand factor concentrate are usually given to patients with aVWS and active bleeding. In addition, platelet transfusion might be considered in bleeding complications if an underlying platelet dysfunction is suspected.53
The data summarized here reflects the need for an individualized assessment of the bleeding/thrombosis risk in each patient and emphasizes the need for tailored therapeutic decisions.
THROMBOTIC COMPLICATIONS
Patients with MPNs have increased risks of thrombotic complications compared to age- and sex-matched individuals without MPNs. The occurrence of thrombosis is one of the main contributors to the increased morbidity and mortality in MPN patients. Thrombosis can be one of the initial manifestations that lead to the diagnosis of MPN.45,60 The reported prevalence of thrombosis among patients newly diagnosed with MPN varies considerably across studies.46–50 A meta-analysis, including 29 cohort studies with 13,436 MPN patients, showed that the pool prevalence of thrombosis was around 20%.51 The mechanisms associated with the occurrence of thrombosis in MPN patients are considered to be multifactorial; indeed, many aspects remain insufficiently clear and biological studies frequently show contradictory data. Uncontrolled cytosis, cell proliferation and cell activation of any hematopoietic lineage, exacerbation of underlying inflammation; changes in the coagulation system, abnormal cell adhesion, as well as participation of vascular endothelial factors are postulated to be participating factors.61 Importantly, whereas erythrocytosis62 and leukocytosis63–66 have been associated with thrombosis in several studies, thrombocytosis appears not as strongly correlated with the thrombotic risk.64 However, the increased thromboxane synthesis observed in PV platelets67 suggests that thromboxane-dependent platelet activation contributes to the increased risk of thrombosis among PV patients.
A retrospective study of 1,537 Chinese MPN patients showed that presence of a JAK2V617F allele burden above 50% was significantly more frequent in patients with thrombosis compared to those without thrombotic complications.68 Thrombosis in patients with MPN could present as mild microcirculatory disturbance or as major arterial and venous thromboembolic events such as ischemic stroke, myocardial infarction, peripheral arterial diseases, and venous thromboembolism.69 MPN patients are at not only a high risk of developing arterial and venous thromboembolism but may develop as well thromboses at unusual sites such as the hepatic, portal, and splenic veins, the cerebral sinuses, and the mesenteric arteries. Patients with splanchnic vein thrombosis or cerebral sinus vein thrombosis may lack myeloproliferation but carry already MPN driving mutations at diagnosis. JAK2V617F mutation is the driving mutation, which is the most frequently associated with unusual site thrombosis.70–74 Testing for JAK2V6127F mutation is therefore usually performed in patients with splanchnic vein thrombosis, and testing for CALR may be restricted to JAK2V617F-negative patients with a spleen size of at least 16 cm and a platelet count above 200×109/L.75
Patients may be assigned to a thrombotic risk category depending on the patient- and disease-related variables.76 Accordingly, the thrombotic risk has an important weight on therapeutic decisions in patients with PV and ET. On the contrary, in MF, the thrombotic risk is not consistently included in the therapeutic algorithm. In PV, age and history of previous thrombosis are the prognostic factors used to classify patients in low-risk and high-risk categories. In ET, the international prognosis score of thrombosis (IPSET) system is the recommended prognostic system comprising age, previous thrombosis, cardiovascular risk factors, and JAK2V617F mutation.77–80 It should be applied to all ET patients at diagnosis. Furthermore, control of general risk factors for thrombosis, including smoking habits, diabetes mellitus, arterial hypertension, and hypercholesterolemia, is as well recommended.81
MYELOPROLIFERATIVE NEOPLASMS DISEASE TRANSFORMATION
MPNs can evolve into fibrotic forms or have a clonal evolution transforming into more serious myeloid diseases such as acute myeloid leukemia (AML).82–85 Some MPNs like ET show a more benign course and transform less frequently; however, fibrotic post-ET has been described. Previously, many ET patients had no BM histological evaluation at time of diagnosis. Thus, we may hypothesize that some of these patients were incorrectly labeled as ET despite having, probably, a pre-fibrotic form of MF instead. The most recent WHO updates8 suggest systematic BM trephine biopsy investigation at first presentation. This will allow a better estimation of the real risk of fibrotic transformation of ET in the future. Long-lasting PV may progress to a spent phase, called post PV-myelofibrosis; clear risk factors for this progression are not well defined. Myelofibrosis as an entity per se, may initially present in an early form, with little evidence of fibrosis, and progress over time into an overt form.
MPNs can progress to secondary AML. The recognition of potential risk factors for such a dramatic transformation is of great interest. It is being considered that this risk is influenced by factors related to the disease and the treatments received as well. A nationwide MPN cohort study evaluating 11,039 patients in Sweden showed that the risk of AML/MDS development after MPN diagnosis was significantly associated with high exposures of Phosphorus-32 (32P) and alkylators but not with hydroxyurea (HU) treatment. Likewise, this study showed that 25% of MPNs patients who developed AML/MDS were not exposed to cytotoxic therapy, supporting a significant role for factors without relation to therapy.86 Postulated disease-related factors include mutations of CALR, splicing factors, epigenetics, and signaling molecules of RAS proteins.87–89 A recent study evaluating the long-term outcome of 3,023 consecutive MPN patients showed a leukemic transformation in 6%. The incidence of leukemic transformation was significantly higher in patients with PMF compared to those with PV (9.3% vs 3.9%; P<0.001) and ET (9.3% vs 2.6%; P<0.001) but remained comparable in patients with PV versus those with ET (P=0.22).85
MOLECULAR INVESTIGATIONS IN MPNs
ROLE OF DRIVER MUTATIONS FOR PRECISION MEDICINE
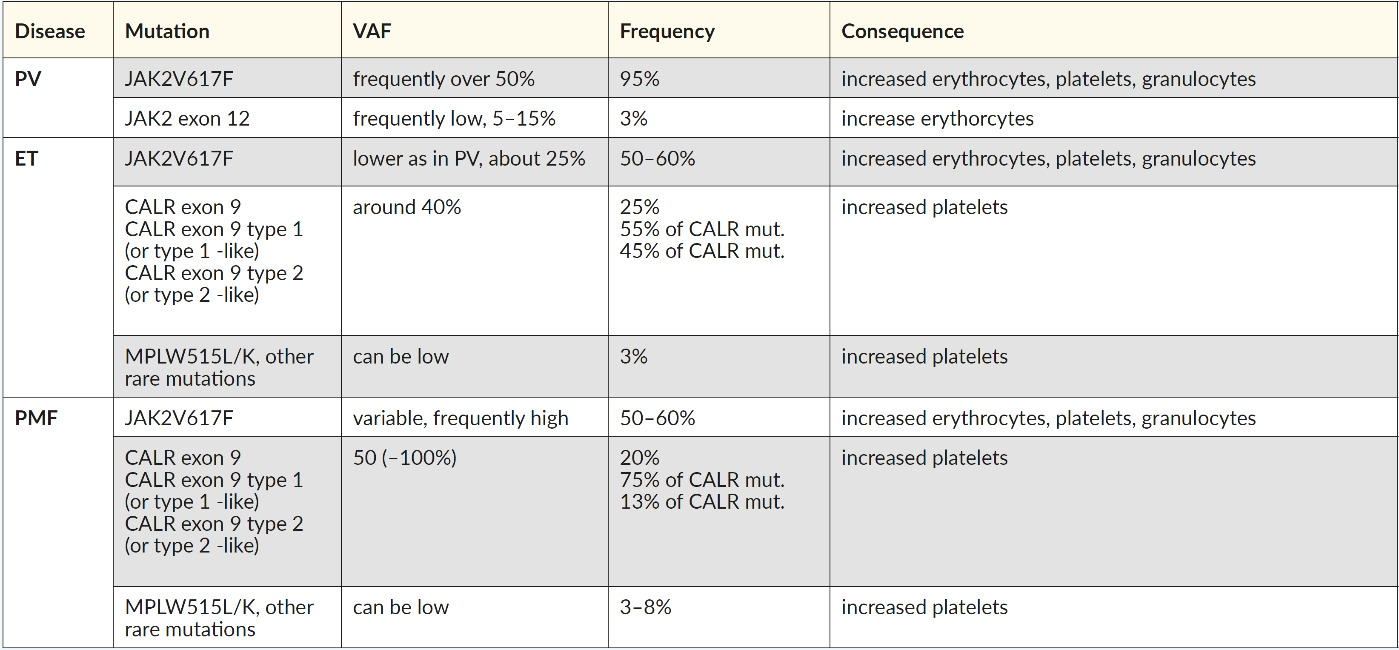
Already in 1960, the Philadelphia chromosome was discovered by Hungerford and Nowell, discriminating CML from the other, BCR-ABL1 negative MPNs being discussed in this chapter.90 However, more than four decades passed by before the JAK2V617F was detected in PV.91,92 This was followed in 2006 by the identification of the next genetic variant: MPLW515L and W515K.93 The next molecular discovery followed in 2013 by two separate research groups: CALR mutations in exon 9, leading to a frameshift resulting in a novel C-terminal peptide sequence.15,94 All three mutations JAK2, MPL and CALR, affect the JAK-STAT pathway, the principal signaling mechanism for a wide array of cytokines and growth factors (Figure 1). JAK2 mutation activates signaling through MPL (involved in erythrocytosis), EPOR (thrombocytosis), and G-CSFR (neutrophilia) cytokine receptors. Loss of heterozygosity is quite frequent, leading to very high variant allele frequency (VAF) up to 90–100%, particularly in PV.

CALR exon 9 frameshift mutations lead to activated MPL and less markedly G-CSFR, resulting in thrombocytosis but not erythrocytosis. Eighty percent of mutated CALR belong to one of the two following types: a 52 bp deletion, named type 1, and a 5 bp insertion, named type 2 mutation. The remaining 20% of cases are classified as type-1, respectively type 2- like mutations, depending on their conformation.
MPL mutations lead to dimerization of the MPL receptor, which in turn causes a constitutional activation of this receptor, with similar results as JAK2V617F.
In general, JAK2 and MPL mutations have the least positive effect for the patients, followed by CALR type 1-like mutations, whereas the best outcome is observed for CALR type 2-like mutations.
Routinely, molecular investigations are done testing sequentially, beginning with the most common MPN mutations according to their frequency: JAK2V617F, CALR exon 9, and MPL, followed by the analysis of JAK2 exon 12 for PV. The frequency of the common driver mutations and variant allele frequencies can help to distinguish PV, ET, and PMF (Table 2). Patients who carry none of these three mutations are classified as “triple-negative” patients, for whom a more in-depth screening is needed for diagnosis, prognosis, and therapy decisions.
EXTENDED PANELS CONTRIBUTING TO RISK STRATIFICATION
Risk stratification is a key tool for decision-making in any disease with markedly heterogeneous outcomes. In this regard, investigations of additional mutations have contributed enormously in helping to identify high-risk patients. Patients with severe forms of the disease may be cured by allogeneic hematopoietic stem cell transplantation (HSCT), it would therefore be mandatory to identify them early enough not to lose timely this unique therapeutical option. In PMF, this is the case, risk stratification needs a workable scoring system that is as precise as possible but remaining reasonably simple to handle.
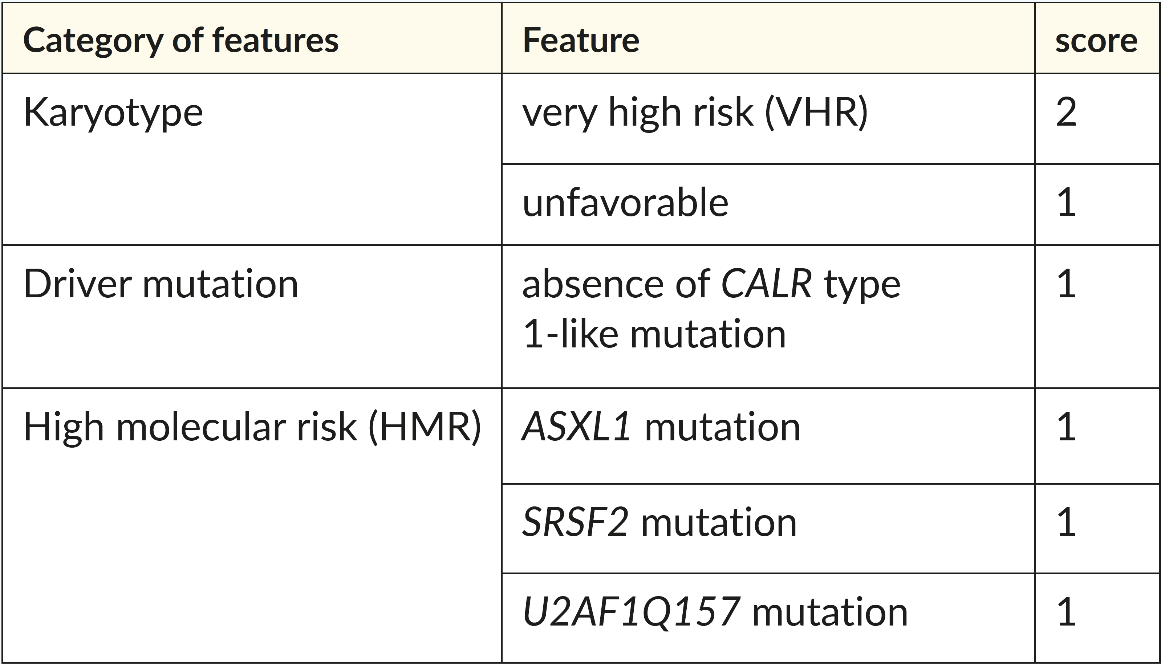
For PMF, international collaborations have introduced prognostic scoring systems since 2009, which help decision-making regarding treatments like HSCT for high-risk patients. The first system was based on clinical variables.77 Subsequently, MIPSS70 and MIPSS70-plus added genetic variables.28 In recent years, several scoring systems with diverse molecular and clinical features were published. In 2018, a new tool was developed,96 which relies solely on cytogenetic and molecular markers: the genetically inspired prognostic system for primary myelofibrosis (GIPSS). The preferred two-step approach of the authors is that the GIPSS is scored first, stratifying the patients into four prognostic groups (low, intermediate int1, int-2, high risk) (Table 3). Patients falling into the two intermediate categories are further evaluated using the MIPSS70+ scoring system (Table 4), which allows improved discrimination. This approach represents an easy-to-use system for the first evaluation, with the possibility of refining the findings in a second step.


PRECISION GENOMICS
Chronic myelomonocytic leukemia (CMML) provides a good example of the value of precision genomics for prognostication and targeted therapeutic advances based on the implementation of a variety of genetic markers for diagnosis and therapeutic management.97 Currently, also in MPNs, progress is made towards personalized predictions of patient outcomes and optimal therapeutic decisions. In 2018, eight subgroups of MPNs were identified by comprehensive genomic characterization defining distinct genetic subgroups and by integration with clinical variables.89 We can recognize the heterogeneity of MPNs and their prognosis on the combination of the driver and the additional associated mutations.98,99 The most common MPN-related mutations affect epigenetic regulation, splicing, and signaling. All these mutations are associated with a poor prognosis and progression of the disease. Many of these genes, such as DNMT3A, ASXL1, and TET2, are associated with clonal hematopoiesis of indeterminate potential (CHIP) and therefore are found with increasing incidence in elderly patients, often associated with an increased risk of subsequent hematologic malignancies.100 Several studies have shown that the higher the number of adverse mutations is as well for the prognosis significant.98,101
The most commonly mutated non-driver genes involved in epigenetic regulation are from two different groups, with TET2, DNMT3A, and IDH1/2 involved in DNA methylation and ASXL1 and EZH2, being part of the polycomb repressor complex modulating chromatin structure. In up to 22% of MPN cases, TET2 mutations were identified, and patients acquiring the JAK2V617F driver mutation prior to the one in TET2 present with a more severe phenotype and are more likely to be diagnosed with PV compared to ET. Furthermore, they suffer from an increased risk of thrombosis.102 In less than 10% of patients, JAK2V617F is followed later on by a DNMT3A mutation, more likely associated with PV or MF rather than ET.103 Mutations in IDH1/2, EZH2, and ASXL1 are associated with an increased risk of leukemic transformation.104
Mutations in regulators of the splicing machinery generally occur in PMF, mainly affecting U2AF1, SRSF2, ZRSR2, and SF3B1 genes. Rarely, cell signaling (SH2B3, CBL, NRAS/KRAS, and PTPN11) or transcription factors, like RUNX1, are affected. The occurrence of these alterations correlates with poor prognosis and disease progression.98,99 Mutations in DNA damage response genes, particularly TP53, often associated with complex karyotypes or abnormalities in chromosomes 5 and/or 7, are detected at later stages of the disease and correlate with progression or can be related to previous treatments as demonstrated for PPM1D.105
Many additional somatic mutations found in MPNs are already present at diagnosis. Therefore, a comprehensive mutation screening at diagnosis provides considerable prognostic information, including the risk of leukemic progression differing significantly among the entities and mutational profiles that supports therapeutic decisions and thereby is a key factor for the implementation of precision medicine for the treatment of MPNs.
THERAPEUTIC DECISION MAKING IN MPN PATIENTS
At the time of initial presentation, MPN patients may have symptoms or complications requiring urgent therapeutic intervention. Generally, at this early time point, peripheral blood values and patient’s symptoms are the only information available for therapeutic decisions. Cytoreductive therapy or management of complications such as thromboses, or less commonly, bleeding, may be required. Patients presenting with symptomatic polyglobulia should undergo phlebotomies and/or cytoreduction, likewise, microcirculation problems associated with thrombocytosis require cytoreduction. Hydroxyurea (HU) remains the first-line cytoreductive treatment of choice.106 In rare instances, severe life-threatening microcirculation complications due to polyglobulia or thrombocytosis can also be treated by RBC or platelets apheresis, which may be lifesaving in selected cases.107,108
Once the MPN diagnostic workup is completed and the diagnosis is established, the specific approach in each individual patient should be considered. Indeed, there is no one way to treat all MPNs, each entity requires specific monitoring and treatment according to the prognosis. Many international guidelines for the therapeutic management of MPNs are available.11,81,109 Treatment consists of alleviating systemic symptom burden, controlling disease-proliferation and/or splenomegaly, and prevention of complications while prioritizing patients’ quality of life in each decision. Control of general risk factors for thrombosis, including smoking habits, diabetes mellitus, arterial hypertension, and hypercholesterolemia, is in all MPNs recommended.
The prognostication systems specifically designed for each MPN entity can guide the treatment steps. The thrombotic risk has an important weight on therapeutic decisions in patients with PV and ET. On the contrary, in MF, the thrombotic risk is not consistently included in the therapeutic algorithm. In PV, age and history of previous thrombosis are the prognostic factors used to classify patients in low and high-risk categories. Low-risk patients will be mainly managed with phlebotomies and low-dose aspirin, while high-risk will additionally require cytoreduction.11 In ET, the IPSET is the recommended prognostic system comprising age, previous thrombosis, cardiovascular risk factors, and JAK2V617F mutation (Table 5). Low-risk patients are observed or treated with low doses of aspirin, while high-risk patients will additionally require cytoreduction.77

In MF, there are several models to identify prognosis for treatment decisions. Older age, the presence of anemia, leukocytosis, thrombocytopenia, circulating blasts, constitutional symptoms, need for transfusions and bone marrow fibrosis are factors associated with poor prognosis. Furthermore, cytogenetics features, the type of driver mutations, and high-risk mutations contribute in different scoring systems (Table 3 and Table 4).96 When molecular information is limited, it will be necessary to apply scores that consider only clinical parameters.110,111 In MF, the treatment primarily aims at controlling symptoms, reducing splenomegaly, and fibrosis. Transplant eligible patients must be identified timely; frequently, anti-JAK2 compounds will be used before transplantation.
In general, in MPNs when cytoreduction is indicated, HU, anagrelide, interferons, ruxolitinib, and fedratinib are options for everyday use with different indications.81 A number of ongoing studies investigating different drugs in the field of MPNs anticipate an exciting future landscape in the management of these diseases.112
The treatment of MDS/AML transformed from MPN is difficult; prognosis is in general adverse.113 The commonly advanced age of the MPN population and their comorbidities frequently represent treatment limitations. Chemotherapy-based schemata for AML represent the standard therapy in fit patients; however, many patients will just receive palliative treatments.114 Advances in targeted therapies for AML represent an attractive choice in a field with otherwise few options. HSCT is considered the only curative potential treatment in patients younger than 65 years or as well for older patients still in good general condition.115,116
CONCLUSIONS
Although classic BCR-ABL1 negative myeloproliferative syndromes are a group of diseases with some common aspects, it is extremely important to establish the precise diagnosis of the different entities that compose this group since PV, ET, and MF have different risks, prognosis and require different therapeutical management. Undoubtedly, in recent years, there has been an enormous contribution in the understanding of the molecular aspects of these diseases; likewise, the update of the WHO was an important step in their diagnostic precision. A deep understanding of the laboratory, clinical and complication profile of each entity are valuable contributors for greater accuracy in patient management.
TAKE-HOME MESSAGES
-
In MPN negative for BCR-ABL1, consideration of individual variability is imperative in both diagnosis and treatment of the disease.
-
The combination of clinical, morphological, and molecular genetic characteristics is the most appropriate attempt to define MPN.
-
Bone marrow morphology remains the central hallmark in the 2016 WHO MPN classification.
-
Prognostic systems designed specifically for each MPN entity will guide the steps of treatment.
CONFLICT OF INTEREST
LN: none
NP: none
VUB: none
AAS: Consultancy Vifor Pharmaceuticals
AR: Novartis: Consultancy, Honoraria, Advisory Board, Research Funding; CSL Behring: Research Funding; Alexion: Research Funding; Orphaswiss: Advisory Board. Celgene: Advisory Board Consultancy. AstraZeneca: Advisory Board.
AUTHOR’S CONTRIBUTIONS
Linet Njue (LN), Naomi Porret (NP), Vera Ulrike Bacher (VUB), Anne Angelillo-Scherrer (AAS) and Alicia Rovó (AR). LN, NP, AAS, VUB, and AR wrote the manuscript. LN, NP, and AR created the tables and figures. All authors revised the manuscript critically and approved the final version.