INTRODUCTION
For a long time, docetaxel has been the only life-prolonging therapeutic option in metastatic castration-resistant prostate cancer (mCRPC).1 Fortunately, our armamentarium has evolved since the beginning of this decade, with positive results from large phase III trials every year, which led to the approval of cabazitaxel, abiraterone, radium-223, enzalutamide and apalutamide. However, the prognosis could be improved not only through new drugs, but also by a redefinition of the primary metastatic hormone-sensitive space and the early combination of androgen deprivation therapy (ADT) with these drugs as well as local interventions like radiotherapy (RT).2,3
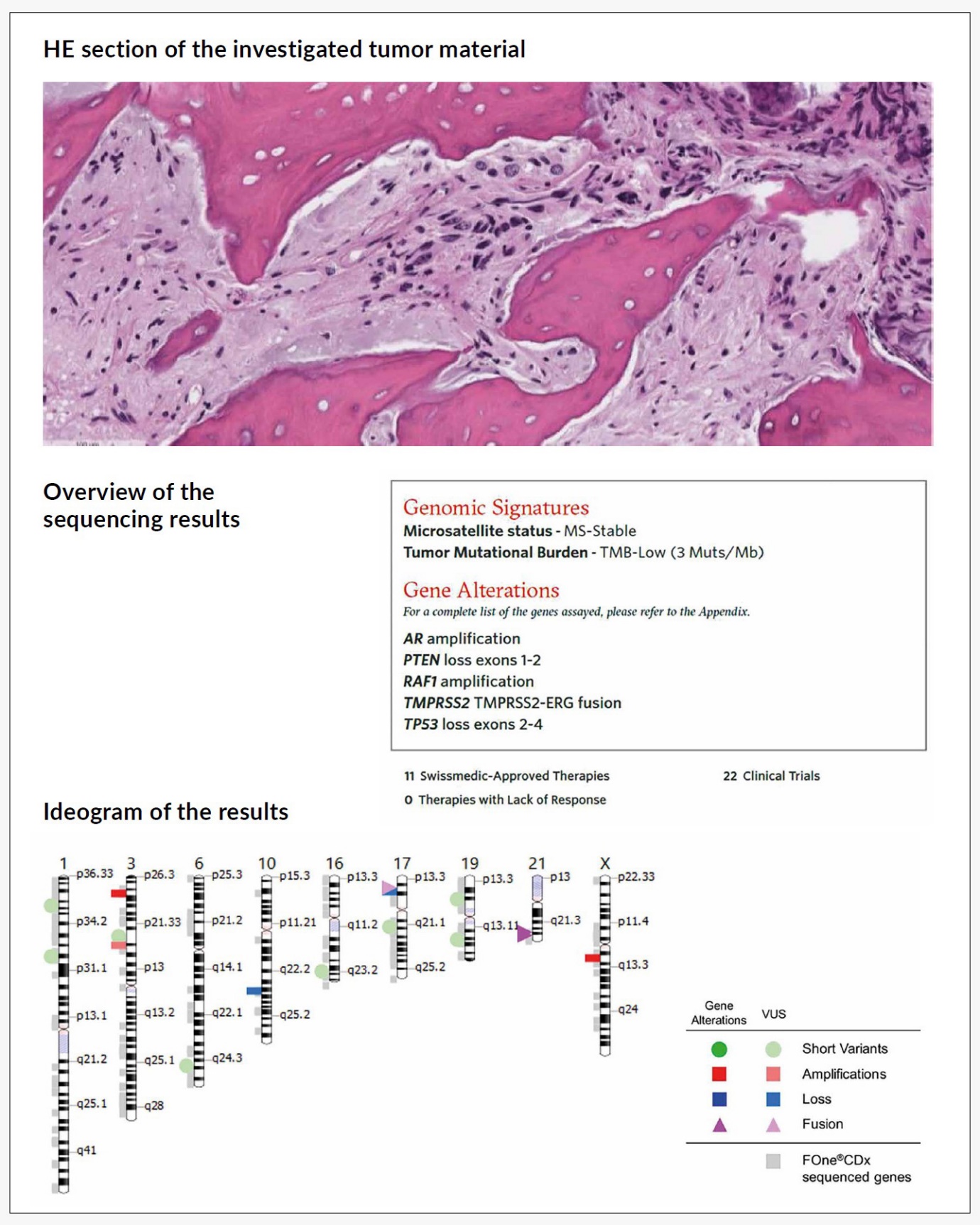
Nevertheless, the majority of patients will end up in a situation, where the standard of care (SOC) treatment options have been exhausted and new therapeutic targets are urgently needed.3 This ultimately happened to the mCRPC patient we presented in the last issue of healthbook TIMES Oncology Hematology.4 When the patient experienced the fifth disease progression after local RT, docetaxel, cabazitaxel and enzalutamid, a computed tomography (CT)-guided re-biopsy and next-generation sequencing were performed (Figure 1).

GENOTYPE-DRIVEN APPROACHES IN METASTATIC CANCER
New sequencing technologies have enabled the rapid and cost-effective detection of gene alterations in cancer. The initial efforts have focused on primary tumors,5,6 but recent investigations have extended this approach to metastatic cancers,7–9 which are responsible for more than 90% of cancer-related deaths. These analyses have shown a high consistency in spatially and temporally heterogenous samples. Nevertheless only a minority of patients have received a genome-guided therapy, which might be explained by reduced performance status, comorbidities, limited access to clinical trials and alternative options or patient preferences.10,11
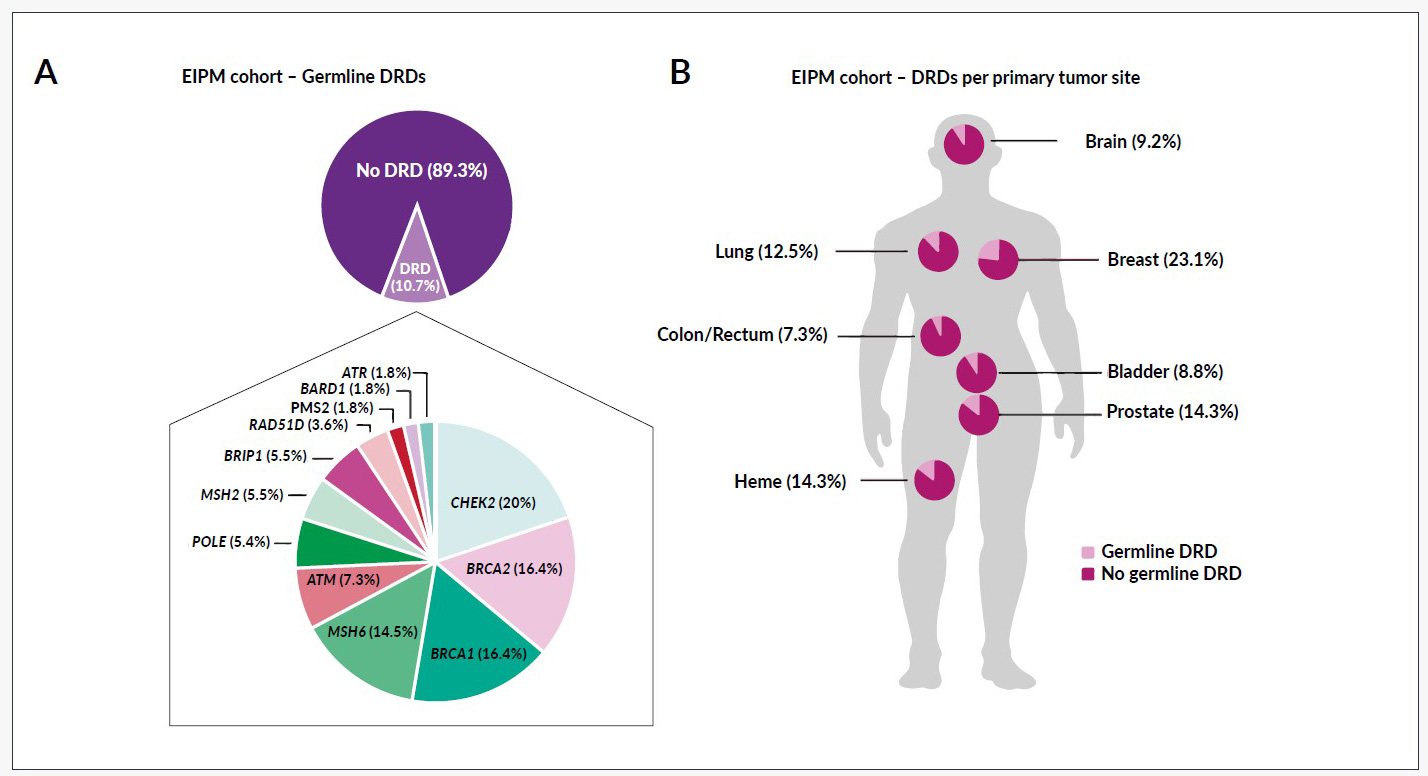
Apart from the low rate of trial enrollment, the genomic analysis of metastatic cancers has shown an incidence of germline DNA repair defects in around 10% of the patients (Figure 2), irrespective of the tumor entity (7.3–23.1%).7 Besides the therapeutic implications, these results impact genetic counseling, familial screening and primary prevention in healthy individuals who carry the mutation.

GENOMIC TESTING IN PROSTATE CANCER
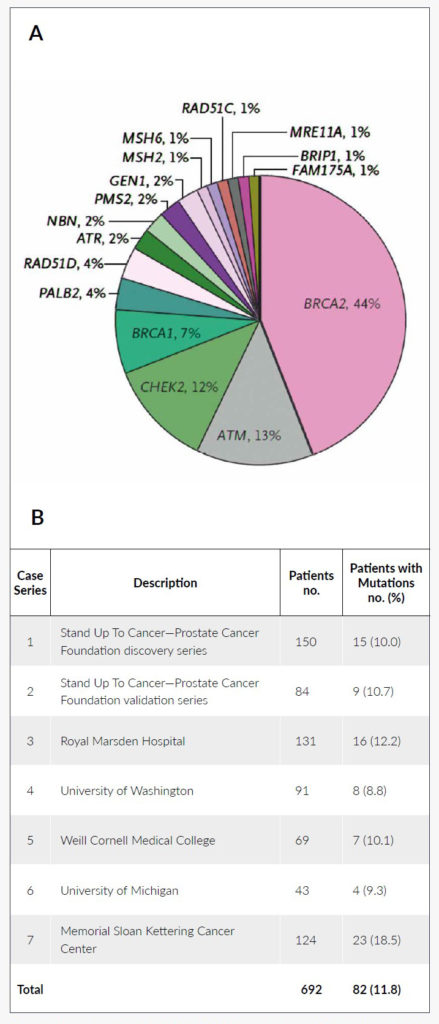
Although mutations in the BRCA1/2 genes have been described earlier, the frequency has been underestimated in these analyses (BRCA1: 0.44% and BRCA2: 1.2%).12,13 Later investigations have shown a markedly higher rate of mutation carriers and could associate germline BRCA1/2 mutations to a higher incidence of disease, more advanced disease at diagnosis and poorer outcomes.14 When patients with primary metastatic disease were analyzed, the detection rate was even higher and other than BRCA1/2 mutations, genomic alterations could be found in 10 additional genes (Figure 3).15 Interestingly, half of the patients in this cohort did not report any family history of cancer.

To create the framework for precision oncology in advanced prostate cancer, several other trials have used whole-exome and transcriptome sequencing to identify mutational landscapes in mCRPC and uncover resistance mechanisms to current standard treatments.17–20 Robinson et al. (2015), for example, detected the most frequent alterations in the androgen receptor (AR) gene, including splice variants (63%), TP53 (53%), PTEN (41%) and ETS rearrangements (57%).17 Compared to primary prostate cancer, AR - and TP53 alterations were found to be enriched in mCRPC cases. DNA repair gene alterations showed an even higher frequency (22.7% overall and 8% germline mutations) than previously reported. Taken together, these analyses were able to find actionable mutations in 89% of the mCRPC patients and uncover important resistance mechanisms, which can potentially influence clinical decision making.
PREDICTIVE AND PROGNOSTIC VALUE OF AR PATHWAY ALTERATIONS
As mentioned above, AR gene alterations are the most frequent in mCRPC (63%). They include copy number gains, point mutations and splice variants.17,18,21 When mutations of FOXA1 (pioneer transcription factor), NCOR1/2 (negative regulator of AR) and SPOP (transcriptional regulator of AR) are taken into consideration, AR pathway alterations affect nearly 3 out of 4 men with mCRPC.17,22
When AR gain or AR mutations (nonsynonymous) were detected in patient plasma circulating tumor (ct) DNA, it was 5 times less likely to have a prostate-specific antigen (PSA) response (PSA decline ≥50%) on abiraterone. These AR alterations were also associated with a detrimental progression-free survival (PFS) (HR: 3.73 [95% CI: 2.17–6.41]; p<2 x 10-6) and overall survival (OS) (HR: 7.33 [95% CI: 3.51–15.34]; p<1.2 x 10-7). Similar observations have been made with the AR splice variant 7 (AR-V7), a short form of the AR, which lacks the N-terminal domain and is constitutively active without ligand binding. When treated with enzalutamide or abiraterone, patients with AR-V7 in circulating tumor cells (CTCs) showed a markedly reduced PFS (1.3–1.4 months) and OS (5.5–10.6 months).
No mCRPC patients responded to both AR targeting agents with a detectable AR-V7 compared to more than half of the patients when AR-V7 was undetectable (53% for enzalutamide and 68% for abiraterone).21 Due to their retrospective nature and the lack of reproducibility (partially explainable by methodological differences), these results have raised countless critical discussions. Eventually, the PROPHECY study allowed a prospective validation of the initial results.23 Recent investigations have also shown that AR-V7 is a treatment-emergent event after ADT (<0.1% in primary prostate cancer vs. 75% of the cases after ADT) and that its expression increases further after exposure to novel AR targeting drugs.24 Both trials have confirmed the negative impact of AR-V7. The authors suggested that patients should be offered alternative treatments. In the presence of AR-V7, taxane chemotherapy seemed to provide an improved PFS (HR:0.19 [95% CI: 0.07–0.52] for PSA PFS, p=0.001; HR:0.21 [95% CI: 0.07–0.59] for PFS, p=0.003) compared to enzalutamide or abiraterone.
Taxane chemotherapy was also associated with higher response rates (41% vs 0%; p<0.001). Moreover there seemed to be no negative associations between AR-V7 positivity and PSA response, PFS or OS.25 Nevertheless, it should be mentioned that the trial cohort was relatively small (n=37) and the OS was poor irrespective of the treatment. Another study investigated the influence of plasma AR gain determined by droplet digital polymerase chain reaction (ddPCR) on the efficacy of cabazitaxel and reported a shorter OS and PFS (OS 10.5 vs 14.1 months, HR: 1.44 [95% CI: 0.98–2.13], p=0.064; PFS 4.0 vs 5.0 months, HR: 1.47 [95% CI: 1.05–2.07], p=0.024). In an exploratory analysis, the effects seemed to be even more pronounced in patients with an initially reduced dose of cabazitaxel (OS 7.3 vs 14.1 months, HR: 1.95 [95% CI: 1.13–3.83], p=0.016; PFS 2.7 vs 5.0 months, HR:2.27 [95% CI: 1.39–3.71], p=0.001).
In contrast, speckle-type POZ protein (SPOP) mutations represent a class of mutations which are associated with CHD1 loss and an increased sensitivity to abiraterone.26 They are not prognostic and occur early in around 10% of prostate cancer patients. SPOP mutations are associated with a longer median treatment duration of abiraterone (HR: 0.37, p=0.002) and increased OS from the start of abiraterone treatment (35.0 vs 14.3 months, p<0.001).
CLINICAL IMPLICATIONS OF DNA REPAIR DEFECTS IN PROSTATE CANCER
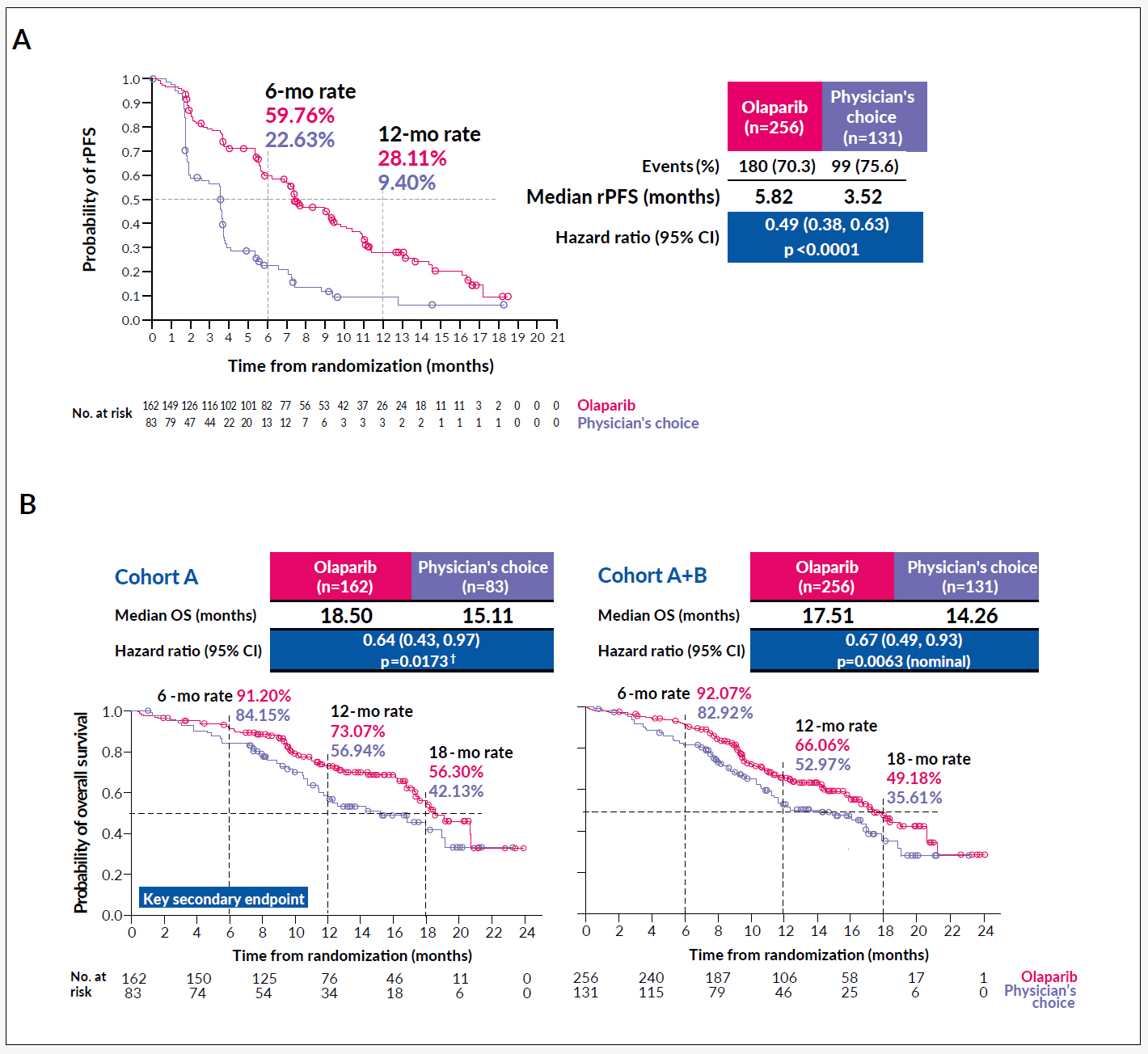
Evidence from ovarian, fallopian tube, peritoneal and breast cancer suggest that mutations in homologous recombination repair (HRR) genes (e.g. BRCA1/2, PALB2, ATM, RAD51C/D or CHEK2) can predict response to platinum-containing chemotherapy and poly(ADP-ribose) polymerase inhibitors (PARPi).27,28 Such exceptional responses could also be shown in mCRPC patients harboring homologous DNA repair defects – for both carboplatin and olaparib (PARPi), although many patients were heavily pre-treated.16,29 Based on these findings, the PROfound phase III trial (NCT02987543) was initiated, which compared the efficacy of olaparib with abiraterone/enzalutamide (physician’s choice) in molecularly preselected patients with defects in any of 15 prespecified HRR genes. Cohort A included patients with BRCA1/2 or ATM mutation, while patients in cohort B had one of the other 12 mutations (BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D or RAD54L). Overall, 4425 patients were screened (Foundation One Inc.) and they were randomized in a 2:1 ratio (n=265 in cohort A and n=142 in cohort B). The initial results have been presented during this year’s ESMO meeting. The study was able to reach the primary endpoint of a prolonged radiographic PFS (rPFS) and showed a trend towards improved OS besides a cross-over rate >80% (Figure 4).30

Cyclin-dependent kinase 12 (CDK12) bi-allelic mutations31 and microsatellite instability (MSH2 and MLH6)17,32–34 are rare in mCRPC, but have been associated with a higher tumor mutational burden (TMB), increased lymphocyte infiltration and improved sensitivity to immune-checkpoint inhibitors (ICIs) (54.5% with a PSA ≥50% decline and 36% radiographic responses/partial remissions).33 Such responses are otherwise uncommon in an unselected patient population (17% PSA responses ≥50% and 8% PSA responses ≥90%).35,36
ACTIONABLE POTENTIAL OF OTHER GENOMIC ALTERATIONS IN PROSTATE CANCER
The PI3K/mTOR pathway plays an important role in mCRPC. Various alterations do not only affect half of the mCRPC cases (PTEN 40.7%, PI3KCA 5.3%, PI3KCB 6.0%, PI3KR1 5.3%, AKT1 1.3% – altogether 49% with partially overlapping events), but also have a negative prognostic impact on various treatments.37,38 A phase II trial (NCT01485861) suggested that the combination of the PI3K inhibitor ipatasertib plus abiraterone is superior to abiraterone alone, especially in mCRPC with PTEN loss (rPFS 11.5 months vs 4.6 months, HR: 0.39 [90% CI, 0.22–0.70]). Therefore, the phase III trial IPATential 150 (NCT0307228) has been initiated.
TP53 (53%) and RB1 (9%) harbor additional genetic alterations, which are associated with CRPC and are amongst the strongest negative outcome predictors for the treatment with novel anti-androgens.39–42 While drugs that target mutant p53 are still under investigation,43 Rb1 inactivation might play a role in the selection of the available drugs, as they might sensitize the tumor cells to cabazitaxel.44 In mouse models, the combined inactivation of Rb1 and p53 leads to increased lineage plasticity by upregulated expression of epigenetic reprogramming factors like Sox2 and Ezh2, which resembles human neuroendocrine prostate cancer.45–47 But inactivation of SOX2 with short hairpin-RNA (shRNA)46 or pharmacological inhibition of Ezh2 (GSK126/EPZ6438)47 re-sensitize the tumor cells to enzalutamide treatment. Although it is widely used, it is not yet clear, if these neuroendocrine variants benefit from chemotherapy combinations (carboplatin + taxane or etoposide),45 due to the lack of randomized clinical trials. A recent investigator-initiated trial (NCT01505868), which excluded neuroendocrine/small cell variants has at least shown an improved tumor control with carboplatin plus cabazitaxel compared to cabazitaxel monotherapy (median PFS 4.5 months to 7.3 months; HR: 0.69 [95% CI: 0.50–0.95], p=0.018) for other aggressive mCRPC variants. However, this analysis was not based on the evaluation of OS.48
DISCUSSION & OUTLOOK
The above results make it difficult to agree to critical comments stating that precision medicine is an illusion - especially in prostate cancer.49 89% of the actionable mutations speak their own language.17 But also in other tumor entities, like acute myeloid leukemia (AML), non-small cell lung cancer (NSCLC) or metastatic colorectal cancer, precision oncology has rather become a clinical reality and is not an unfulfilled hope. Nevertheless, the concept of “one mutation one drug” might be too simplistic (e.g. palbociclib in CDKN2A altered pancreatic cancer),50 although it was applicable in the past for cancers with a strong oncogene addiction like EGFR mutant (EGFR M+) NSCLC or chronic myeloid leukemia (CML).51
How to obtain the genetic information (biopsy or liquid biopsy) and how to address tumor heterogeneity,52,53 remain important and yet unanswered questions. The problem of clonal hematopoiesis adds additional complexity. Some of the latest research has shown that it is not enough to prove the pure existence of genetic alterations, but that these tests (AR gain in this case) need further refinement and cut-offs like any classical laboratory test.54 We also have to face the challenge of combining the latest advances in imaging (e.g. 68Ga-PSMA-PET/CT) and genetics to develop the best and most rational strategies in the future. Initial investigations have indicated an association between a high prostate-specific membrane antigen (PSMA) expression, castrate resistant disease and the occurrence of DNA repair defects.
The success of a pan-cancer precision oncology approach will also depend on the development and accessibility of new drugs. In this context, AMG 510, a KRAS(G12C) inhibitor is a good example demonstrating that it is possible to target, what was considered as untargetable for a long period of time.55 In addition to a tumor control rate of 100% (NCT03600883),56 AMG 510 has proven that the immune microenvironment is influenced by specific mutations on the one hand and that their inhibition can trigger tumor immunity on the other hand. Similar observations have been made in mCRPC,57 which opens the possibility to indirectly target the tumor or immune microenvironment, in case direct inhibitors are not available.
The phase III PROfound and phase IV CARD trial have fundamentally impacted the treatment selection and sequencing in mCRPC.30,58 This study ended years of wild speculations on whether one sequence might be better than the other. The result should remind us that we tend to follow the principles of evidence-based medicine even in the era of precision medicine. It does not mean that we should not think about alternative and innovative trial designs, but we must keep in mind that omitting harm is the first step to the best possible treatment. As old as it is, it remains true: “Primum nihil nocere”!
With the phase III evidence of superiority of olaparib compared to novel anti-androgens in patients with defective HRR, it seems to be impossible to neglect the advantages NGS is offering in the management of mCRPC, even though there is the need for significant improvement. Regulatory authorities should be a part of the implementation process and find feasible conditions, which grant early access to novel cancer drugs without negatively affecting the established, stepwise clinical development. Needless to mention, that the algorithms behind the recommendations, which come along with some of the sequencing reports are not perfect yet and would benefit from further adjustments. This would reduce user-bias, misinterpretation and dependency on local knowledge. At this point we should also admit that prostate cancer has the highest incidence among all male-specific cancers. Thus, pharmaceutical companies have more opportunities to receive funding and gain access to patient material, which facilitates extensive pre-clinical and translational research to further pave the way for clinical trials. Probably, it will be significantly harder to implement genome-driven approaches in case of orphan cancers. Basket trials are a valuable strategy, but involve certain risks, when profound biological knowledge is missing.
CONCLUSIONS
The primary question here is if the patient presented in the case report4 benefited from genetic testing. The results (Figure 1) have shown multiple resistance mechanisms against current standard of care treatments (PTEN exon 1-2 loss, AR amplification and TP53 exon 2-4 loss). These compound genetic events are associated with a poor prognosis in the hormone-sensitive as well as in castration-resistant setting and suggest no putative benefit from a re-exposure to one of the previously used antineoplastic agents.59 However, the results indicated a possible benefit from ipatasertib and the CXCR2 antagonist AZD5069 (NCT03177187; clinical trial site in Bellinzona, CH), due to the PTEN loss, but the later was not directly mentioned in the report.57,60 Everolimus (PTEN loss) or sorafenib (RAF1 amplification) have been approved by Swissmedic in other indications and were suggested in the report, but had to be discarded due to very little or complete lack of benefit in phase I/II trials in CRPC.61–64
Ultimately, the failure of fourth-line cabazitaxel with a continuously rising PSA (approx. 400% increase) validated the predictions from the NGS analysis. This itself might not be interpreted as a major benefit of this test, although the avoidance of unnecessary side effects and financial burden is clinically relevant. The NGS test has definitely opened up new therapeutic options for our patients. The integration of such opportunities into individual clinical concepts depends on various factors, like performance status, disease dynamics, co-morbidities, ability to travel, familial support, socioeconomic status etc. This should be considered when NGS testing is discussed with the patient or in the interdisciplinary tumor board.
TAKE-HOME-MESSAGES
-
In primary metastatic prostate cancer, germline mutations have been identified in ~12% of the patients. Lack of family history of cancer does not exclude germline mutations.
-
The phase III PROfound trial has shown a doubling of the progression-free survival and a trend towards an improved overall survival for olaparib vs. abiraterone/enzalutamid in metastatic castration-resistant prostate cancer (mCRPC) patients with homologous recombination repair gene alterations.
-
Certain alterations (e.g.CDK12 bi-allelic mutations and mismatch-repair genes) reflect a highly immunogenic subclass of prostate cancer and have been associated with an increased number of tumor infiltrating lymphocytes and with superior response to immune checkpoint inhibition.
-
SPOP mutations are associated with an improved sensitivity to abiraterone.
-
Androgen receptor (AR) alterations are very common in mCRPC and display an important resistance mechanism to androgen blockade.
COI
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
The author crafted and approved the final manuscript.