
INTRODUCTION
Colorectal cancer (CRC) is the most common gastrointestinal malignancy and a leading cause of cancer-related death.1 By the next decade, the global burden of CRC is anticipated to increase by 60%, with 2.2 million new cases and 1.1 million deaths.2 Typically, CRC patients are diagnosed when they already have advanced metastasis, with the involvement of secondary organs, resulting in a high mortality rate. The liver is the most common site of secondary metastasis.3 Currently, CRC patient prognosis is based on clinicopathological features and largely focuses on the cancer stage at the time of diagnosis. The overall five-year survival rate is approximately 90% for stage I; it declines to 70% for stage II, 58% for stage III, and less than 15% for stage IV.4 Therefore, early detection and intervention, with oncologic resection of the primary tumor, is paramount to improve CRC patient outcomes. Following surgical removal for early and locally advanced CRC, current treatment protocols recommend adjuvant treatment for stage III disease, characterized by lymph node involvement;5,6 adjuvant treatment in stage II CRC shows a limited effect, even in high-risk patients.5–10 Only approximately 20% of stage III patients will fully benefit from adjuvant therapy in the first 3 years, and about 30% will relapse. Therefore, the current treatment algorithm exposes up to 80% of patients to unnecessary toxicity. The IDEA trial, amongst others, highlighted the benefit of 3 versus 6 months of treatment for stage III CRC.10,11 As such, the selection of the most beneficial treatment regimens for CRC patients, especially during the early stages, remains a challenge due to the lack of adequate biomarkers. This topic is currently the focus of several study protocols investigating the impact of ctDNA on therapy decisions after curative resection.12
In an advanced stage, CRC, multi-modality approaches including surgery and systemic treatments with chemotherapy and biological agents such as anti-epidermal growth factor receptor (EGFR) or anti-vascular endothelial growth factor receptor (VEGFR) inhibitors are used in multimodal and multi-sequential steps. One of the major improvements of CRC outcome came by the identification of defined oncogenes in colorectal cancer, which became routine nowadays. Technology advanced quickly in the field of molecular biology, leading to the rapid implementation of DNA testing on tumor tissue samples. Therefore, the presence, or absence, of certain mutations or genes now determines the use of targeted treatments and finally improved treatment selection and outcome of metastatic CRC (mCRC). With the commercial availability of multigene (prognostic/predictive) testing, which can be used in the daily routine, the use of an algorithm to determine relevant predictive and prognostic mutations in mCRC patients at the right time is highly recommended. Prognostic markers provide treatment-independent information on patient outcomes, such as overall survival (OS) and relapse-free survival (RFS). On the other hand, predictive biomarkers support therapy decisions via treatment response information in biomarker-positive patients compared to biomarker-negative patients. In this narrative review, different prognostic and predictive molecular biomarkers currently used in CRC will be discussed. In addition, a new potential biomarker with a possible future role in the surveillance and treatment of CRC patients will be highlighted.
Current biomarker testing in CRC
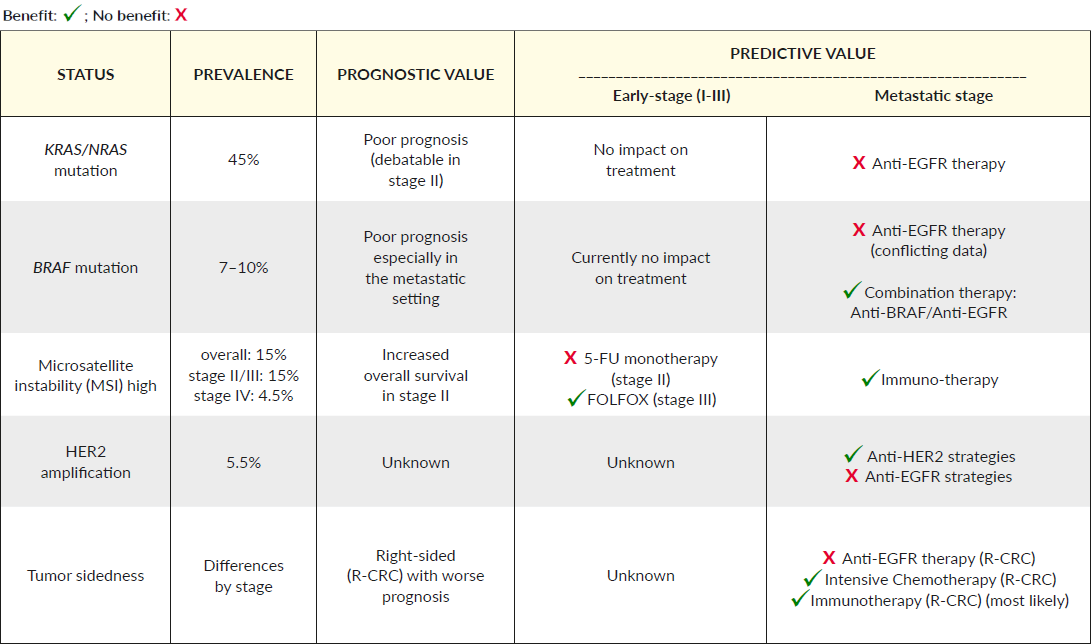
Colorectal cancer is considered as a heterogeneous disease and harbors an accumulation of genetic and epigenetic alterations.13 The majority of CRC tumors are sporadic (70–80%), whereas approximately 20% are hereditary in origin.14 The most common genes mutated in CRC patients are APC (about 82% of cases), TP53 (48–59%), KRAS (40–50%), and PIK3CA (14–18%).15 This mutational status of genes is known to be important in CRC carcinogenesis (e.g., NRAS, KRAS, BRAF) or associated with defects in the DNA mismatch repair system (MMR) (Table 1).
.png)
These defects are the underlying mechanism through which microsatellite instability (MSI) status is determined.16 Recently, new guidelines for molecular biomarker testing in CRC were established to assist in disease prognosis, surveillance, and treatment. The following biomarkers are highlighted for their relevance in CRC treatment.
RAS Mutation
One of the first mutations identified in CRC was KRAS, a downstream effector of the epidermal growth factor receptor (EGFR) and other receptor tyrosine kinases (RTK). In CRC, KRAS mutations are present in 45–50% of metastatic tumors as well as in approximately 15–37% of early-stage tumors.17 KRAS mutations are more often associated with MMR proficient (pMMR) than with MSI tumors. Through epidemiological cohort studies, KRAS mutations were found to predict a worse outcome in CRC patients.18 This prognostic value exists in stage III pMMR, but not if MSI is present.19–21 This information on mutational status and outcome were confirmed by posthoc analysis of data collected from adjuvant clinical studies, including studies of trials PETACC-8 and N0147.19–21
At the start, only KRAS codon 12 mutations (in particular, c.35G>T, also known as G12V), were associated with inferior survival in BRAF wild-type CRC.20–24 More recent data now support the poor prognosis of both frequent exon 2 mutations, including codon 13 mutations.25,26 In conclusion, a 1.5 higher risk of relapse and death was found in KRAS mutant patients compared to KRAS wild-type patients. Moreover, mutations in KRAS and BRAF are associated with inferior progression-free survival (PFS) and overall survival (OS) of metastatic CRC patients in comparison to non-mutated tumors. Several randomized trials showed the prognostic value of KRAS mutations and a total of more than 1200 CRC patients from five randomized trials (FIRE-1, FIRE-3, AIOKRK0207, AIOKRK0604, RO91) were included in the analysis.17 In this meta-analysis, more frequent KRAS exon 2 variants (G12V and G12D) did not have a significant impact on OS. However, the KRAS G12C-variant was associated with a lower OS when compared to the KRAS wild-type tumors (multivariate HR 2.26 (1.25–4.1), p=0.001). A similar trend for inferior OS was observed by the presence of the KRAS G13D-variant (multivariate HR 1.46 (0.96–2.22), p=0.10). At present, a majority of mCRC patients are treated with multi-modality approaches, including surgery and systemic treatments.27 In contrast to chemotherapy without biologics, the addition of an anti-EGFR monoclonal antibody (cetuximab and panitumumab) to the standard chemotherapy regimen has been shown to improve survival, as well as reduce the risk of disease progression.28,29 Due to the constitutive activation of the downstream mitogen-activated protein kinase (MAPK) pathway, this only benefits patient who do not harbor mutations in downstream effectors of EGFR, mainly KRAS and NRAS.30–32
Before starting a systemic treatment, the mutational analysis of KRAS (40% of mCRC cases) and NRAS (7% of mCRC cases) is mandatory prior to treatment with anti-EGFR antibodies.33 The mutational analyses should include KRAS and NRAS codons 12 and 13 of exon two, 59 and 61 of exon three, and 117 and 146 of exon four (5). Importantly, not all wild-type KRAS patients respond comparably to anti-EGFR treatment. Herein the potential emergence of therapy resistance is an important issue. Anti-EGFR therapy leads to the development of KRAS, NRAS, BRAF, and EGFR ectodomain mutations, which stimulates MAPK pathway activation independently of EGFR inhibition. Studies that re-challenged patients with anti-EGFR agents showed an improved overall response rate, most likely because resistant clones decay exponentially after drug removal.34 However, this field is not currently fully understood. Therefore, additional biomarkers are needed for a better selection of that subgroup of the wild-type KRAS population, which is more likely to respond to anti-EGFR therapy, as well as to identify those KRAS wild-type patients who have become resistant.
BRAF Mutation
The BRAF gene is activated by mutations in 10% of CRC patients.35 Therein, mutations most frequently occur in codon 600 (BRAF V600E), which represents over 90% of all BRAF mutations and is typically mutually exclusive with other RAS mutations.36,37 BRAF p.V600 mutational analysis is recommended in patients with pMMR-tumors that display a loss of MLH1, since the presence of a BRAF p.V600 mutation makes Lynch syndrome very unlikely.38 Typically, BRAF-p.V600-mutated tumors are often associated with a higher prevalence in females, higher involvement of positive lymph nodes, high-grade histology, MSI status, and with a higher presentation in the right side of the colon.39 Several retrospective studies underlined that microsatellite stable (MSS) tumors with BRAF mutations have more than two times greater risk of relapse and mortality than those with wild-type BRAF.40–43
The presence of BRAF mutations is associated with reduced survival in stage III and IV, with an objective response rate (ORRs) of <10%, a PFS of about two months, and absolute reduction of OS of four to six months.44,45 This is not the case in stage II, BRAF mutant CRC compared to the BRAF wild-type population.46 Currently, BRAF status testing for stage I and II CRC is not recommended. Until recently, testing the mutational status of BRAF p.V600E has been used exclusively as a prognostic marker for stage III-IV CRC, with little impact on therapy decision. The BEACON study (see below) has now added a predictive element to this setting. Interestingly, the BRAF-CRC subtype should not be defined as one entity as MSI/BRAF-CRC and MSS/BRAF-CRC show different prognoses and outcomes, with a shorter OS and RFS in BRAF/MSS patients and no difference in MSI/BRAF-CRC compared to BRAF wild-type patients.47,48 The response of BRAF mutant tumors to targeted anti-BRAF strategies remains very limited and varies extensively within BRAF V600E cohorts. This heterogeneity in drug resistance might be explained by biologically distinct subpopulations within BRAF -mutated tumors. The future identification of further subgroups of BRAF-CRC might help clinicians to choose more beneficial targeted therapies, as standard treatment regimens are insufficient for BRAF-MSS patients. Several studies demonstrated that BRAF mutations in combination with RAS wild-type lead to a lack of response to anti-EGFR treatments in CRC.49–51 Thus, data concerning the response to EGFR-targeting agents in BRAF-mutant CRCs remain conflicting. As mentioned previously, BRAF inhibition strategies in metastatic CRC with BRAF-mutation have shown a dismal outcome. Recently, pre-clinical studies combining anti-EGFR and/ or MEK or human epidermal growth factor receptor (HER) therapy with BRAF inhibitors demonstrated promising results, which have moved into clinical trials.52–54 Recently, the phase III BEACON CRC study showed that patients with BRAF V600E mutated mCRC benefit from the doublet or triplet chemotherapy-free targeted combination therapy of encorafenib (a BRAF inhibitor), and cetuximab (an anti-EGFR antibody) or combined with binimetinib (a MEK inhibitor) in a second or third-line setting.55,56 Interestingly, at ASCO GI 2020, S. Kopetz demonstrated that the combination of BRAF inhibition with encorafenib and EGFR blockade by cetuximab is sufficient compared to triplet therapy, with a lower toxicity profile. This has led to the Food and Drug Administration (FDA) approval of the doublet therapy as a treatment for patients with advanced BRAF -V600E-mutated mCRC after two prior lines of chemotherapy. Additionally, the ongoing phase II ANCHOR-CRC study (NCT03693170) is investigating the triplet as a frontline treatment for patients with metastatic BRAF V600E-mutant CRC. This approval is a breakthrough for this mCRC population, with a chemotherapy-free targeting combination that will hopefully be directed in the earlier treatment setting in the future. Importantly, given the overlap of BRAF V600E mutations with a high number of MSI, treatment options by checkpoint inhibitors may play an important role in this CRC cohort. Once again, this should be investigated by clinical trials. For the aforementioned reasons, BRAF as a biomarker is recommended by NCCN guidelines in the routine testing in CRC.
MSS/MSI Status
Microsatellites are short tandem repeats of DNA sequences located throughout our genome. Microsatellite instability (MSI) status results from a deficient DNA mismatch repair (MMR) system. Herein, MSI is commonly caused by the inactivation of one of the four MMR genes (MSH2, MLH1, MSH6, and PMS2). Within a deficient MMR system, failure to correct the insertion or deletion of repeating units during DNA replication occurs, which leads to a hypermutable phenotype.
MSI status can be determined by two distinct methods — immunohistochemistry (IHC) analysis or polymerase chain reaction (PCR).57 Reduced expression of the MLH1, MSH2, MSH6, and PMS2 genes, determined by IHC analysis, identifies tumors as microsatellite instable (MSI; also referred to as deficient MMR, dMMR) in contrast to microsatellite stable (MSS; also referred to as proficient MMR, pMMR). Alternatively, standard PCR can be used to compare microsatellite length in tumors versus normal tissue to determine aberrant microsatellite lengths detected in the tumor. As IHC is faster and less expensive, it is commonly used in the daily routine, therefore, PCR testing will be chosen for additional analyses where IHC testing is inconclusive.58 MSI tumors are detected in approximately 15% of all CRC patients, and the prevalence is stage-dependent.47 In stage II/ III CRC, up to 15% are dMMR, whereas only 4–5% of stage IV CRCs are dMMR.59 Of these, 3% are associated with Lynch syndrome, an inherited cancer syndrome associated with a genetic predisposition to CRC, also known as hereditary non-polyposis CRC (HNPCC). The other 12% of MSI tumors are due to sporadic hypermethylation of the MLH1 gene promoter. MSI tumors are more frequently presented in the right colon, are more often associated with younger age, and show poor differentiation with a strong lymphocyte infiltrate. Overall, MSI-high patients show a better prognosis compared to MSS patients.16,38,57 Following this line, the addition of the DNA mismatch repair status to clinicopathological variables has improved prognostic predictions specifically in CRC patients, which is also recommended by ESMO and NCCN.5,28 Hereby, MSI stage II patients have a better prognosis, where additional chemotherapy of fluorouracil (5-FU) shows no benefit, therefore, the recommendation is to place these patients under a surveillance strategy.60,61 The MSI status is less informative in stage III patients, as the risk differences are limited between MSI-high and MSS patients. Moreover, MSI patients seem to have a reduced benefit from 5-FU treatment.62,63 With the new era of immune-oncology and the success of checkpoint inhibitors in different tumor types, such as melanoma and non-small-cell lung cancer, MSI status in CRC patients has become a factor of significant interest for several reasons. Emerging data reflected the higher response of MSI tumors to checkpoint inhibitors, likely due to their higher mutational load and immune cell infiltration.64,65 In 2017, the FDA consequently approved pembrolizumab, a monoclonal anti-programmed cell death (PD) 1 antibody, for use in MSI-high patients, independent of cancer type.66 Additionally, by the positive results of the Checkmate-142 study, nivolumab and ipilimumab are approved for refractory stage IV MSI-high patients.67 Highlighting this, MSI status is the first biomarker-only based indication for therapy, independent of primary cancer (so-called tumor-agnostic approval). Moving the relevance of this biomarker in the earlier stages, MSI status might become a predictive marker for stage III MSI-high patients. New trials have started to test immunotherapy, as a stand-alone or in combination with chemotherapy, in stage III MSI-high CRC (ATOMIC trial, NCT02912559). Unfortunately, not all mCRC patients respond to immunotherapy within the MSI-high subgroup, so further predictive biomarkers are urgently needed to identify intrinsic and acquired resistance to checkpoint inhibition. Of note and in contrast to other tumor types, programmed cell death ligand-1 (PD-L1) expression in CRC tumors did not predict better survival outcomes in patients treated with immunotherapy, which questions the use of PD-L1 as a predictive marker for checkpoint-inhibition therapy in CRC.
HER-2 Status
HER2 (ERBB2) is a transmembrane receptor of the EGFR/ErbB family, and its activation leads to cell proliferation plus inhibition of apoptosis. HER2 overexpression is due to ERBB2 amplification or activating somatic mutations. It is usually defined in clinical practice as IHC 3+ or IHC 2+ and in situ hybridization (ISH)-positive disease. CRC patients presented with a 5% frequency of HER2 overexpression, and about 5.5% ERBB2 amplification.68 With the results of the HERACLES trial (trastuzumab plus lapatinib) and MyPathway (trastuzumab plus pertuzumab), HER2 gained a lot of interest in CRC, as the dual HER2 blockade demonstrated promising clinical benefit with HER2-amplified mCRC.68,69
However, the response rates in mCRC, like in other gastrointestinal (GI) tumors, have not been reproduced with trastuzumab plus lapatinib as seen in the high success of HER2-positive breast cancer. The explanation of these differences is complicated and not currently fully understood. One main fact in CRC might be caused by the heterogeneity of HER-2 IHC staining process and subsequent HER2-positive classification. For breast cancer, complete membrane staining of HER2 is required for a tumor to be considered HER2-positive. In contrast, unlike breast cancers, CRC are gland-forming, mucin-producing tumors with a typical incomplete basolateral and/or lateral staining pattern. When strongly stained, this U-shaped pattern is classified as HER2-positive CRC.70 This intratumoral heterogeneity provides a possible explanation for the differences in the efficacy of the same HER2-targeted therapies between patients with CRC and those with breast cancer. Differences in HER2 expression patterns, and their possible clinical implications, are not limited to CRC as this is also noticed in gastric cancer with negative results in clinical HER2 targeting trials.
There is currently high interest in HER2 as a predictive marker for anti-EGFR therapy. Several studies demonstrated that acquired amplification of ERBB2 negatively predicts efficiency and is associated with the development of resistance to EGFR-targeted therapies.68,69 In summary, and following the ESMO and NCCN guidelines, the implementation of HER2 assessment in daily practice provides useful information for guiding therapy decisions in mCRC.
Tumor localization, Sidedness
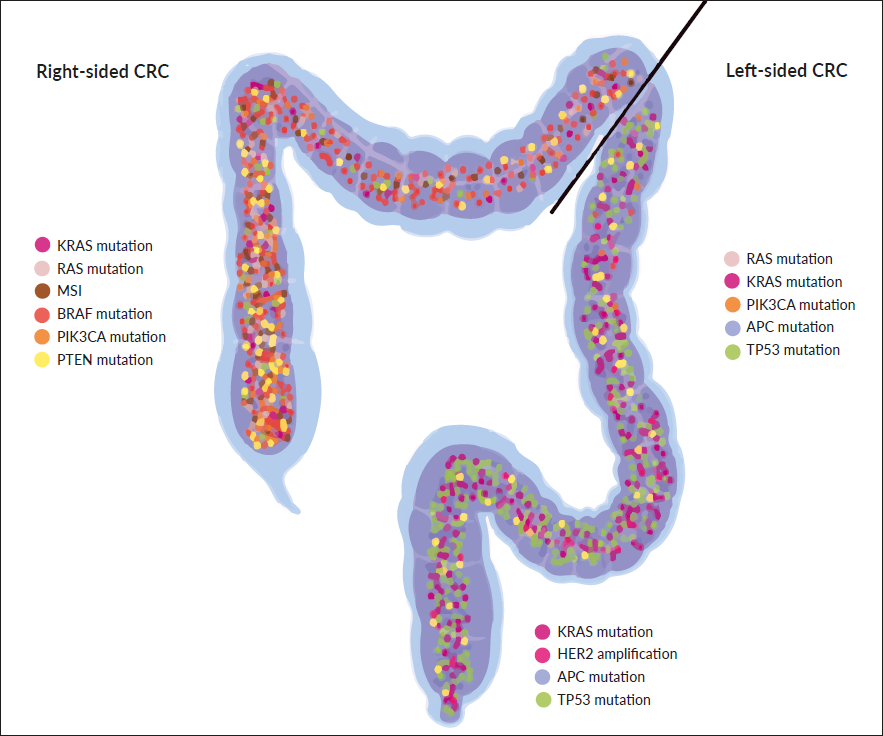
It has been known for decades that the colon has two distinct embryological origins, namely the midgut for the proximal colon (also referred to as right-sided colon) and the hindgut endoderm for the distal colon (also referred to as left-sided colon). Additionally, the two parts of the colon behave differently as they have different blood supplies, distinct microbiome populations, and are characterized by different biological features.71 Several studies support the concept that right-sided colon cancer (R-CRC) shows a worse prognosis than left-sided colon cancer (L-CRC); Figure 1 highlights the different molecular alterations experienced in R-CRC and L-CRC.72,73 Importantly, posthoc analysis of the CRYSTAL and FIRE-3 trials, suggests an association between primary tumor location (PTL) and response to anti-EGFR therapy, as right-sided KRAS wild-type, CRC did not seem to benefit from cetuximab treatment.29 Future study concepts need to analyze PTL as an independent predictive biomarker for anti-EGFR therapy, especially as R-CRC and L-CRC are also characterized by different mutational landscapes. Nevertheless, based on the results, many current guidelines recommend anti-EGFR therapy only to left-sided RAS wild-type CRC.5,27 Finally, due to the high antigenic load, R-CRC suggests more promising results with immunotherapies.71

Promising biomarkers in CRC for later line testing and future aspects
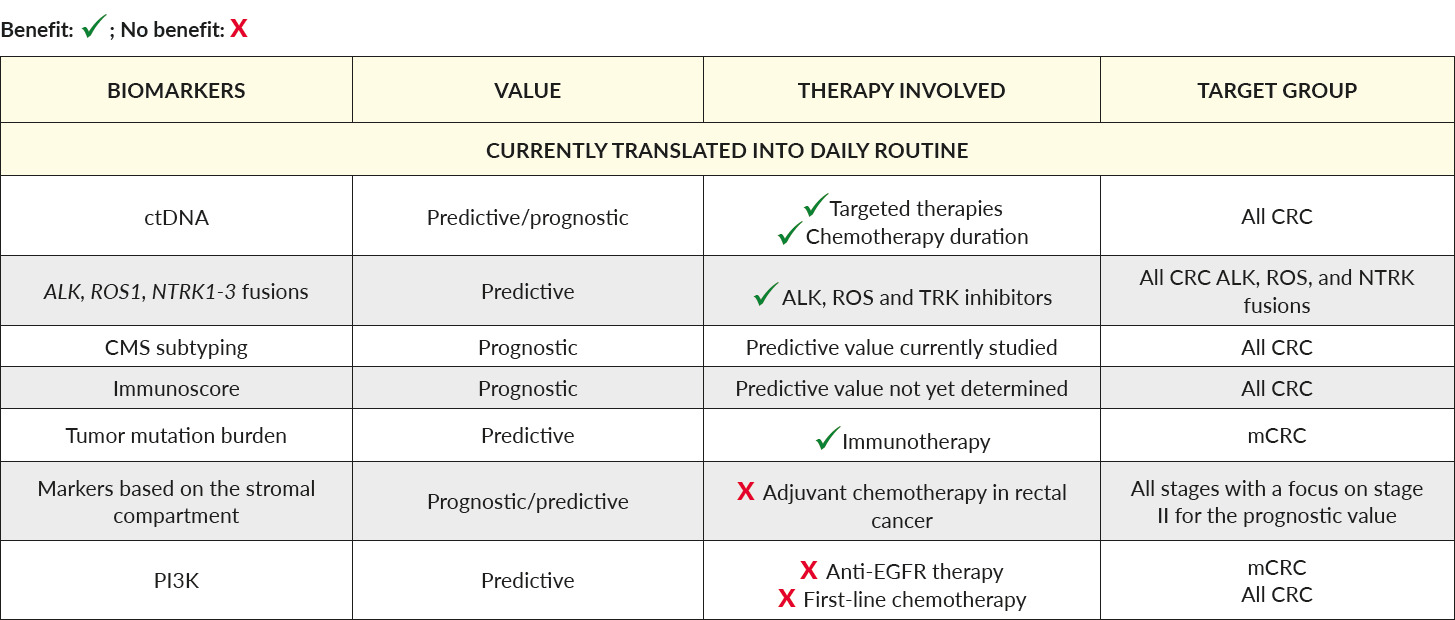
A summary of these promising and future biomarkers is listed in Table 2.
Liquid biopsies, ctDNA, tumor mutational burden and microenvironment
Liquid biopsy may reflect the information of the primary tumor and the metastatic site by the collection and analysis of circulating tumor cells, cell-free nucleic acids, and tumor-derived exosomal vesicles. The identification of ctDNA can be a major challenge: while ctDNA can form up to 50% of total cell-free DNA in later metastatic stages, it can represent less than 1%, or even be undetectable, in earlier tumor stages.75,76 The potential for liquid biopsy for treatment guidance and monitoring the disease is of major interest. Several studies have suggested the possibility of using ctDNA to closely monitor patients after surgery and identify patients with a high risk of recurrence. The independent prognostic value of ctDNA was presented in a recent phase III trial (IDEA-France).77 Following this line, upcoming clinical studies of both early-stage (NCT04120701), and advanced CRC (NCT03844620), will contribute to the impact of this promising diagnostic tool.
Within ctDNA analysis, the tumor mutation burden (TMB) was recently suggested to identify responders to checkpoint blockade therapy.78,79 At this stage, no clear answer to whether TMB is an independent prognostic factor could be made. Moreover, the definition of TMB, as well as the method used to assess it, may vary between laboratories and is not yet clearly defined by NCCN or ESMO. Interestingly, adding MSI and ctDNA to TMB might increase the prediction efficiency of checkpoint inhibitors.80 Further studies involving larger cohorts are needed to validate these findings.
Like in other tumor types, it is increasingly evident that the tumor microenvironment plays an important role in disease progression and tumor resistance. The infiltration of tumors by lymphocytes has been suggested as a prognostic marker.81,82 Based on this observation, the Immunoscore classifier by Galon and collaborators, which assesses the presence of CD3+ and CD8+ lymphocytes within the tumor and invasive margin, was introduced.83,84 Interestingly, while most MSI tumors are infiltrated, the Immunoscore has been reported as a superior predictor than MSI alone.85 However, the efficacy of the Immunoscore in predicting response to immunotherapy agents has not yet been demonstrated so far. Evaluating the impact of tumor stroma is another upcoming field of interest. Herein, tumor-stroma percentage has been confirmed as a favorable prognostic factor in stage II and III CRC (VICTOR trial). Overall survival and disease-free survival (DFS) were significantly lower in CRC with a high percentage of tumor stroma.86 The role of tumor-stroma in the advanced stage is unknown so far.
Rare but clinically relevant mutations in CRC
Overall, the incidence of gene fusion in CRC is rare, ranging between 0.5–2%. However, their prognostic and predictive value justifies to test these markers in the advanced stage after the second line of therapy.87 Highlighting results from the STARTRK study revealed in heavily pretreated mCRC patients harboring LMNA-NTRK1, CAD-ALK, and STRN-ALK fusions that entrectinib, a small molecule which selectively inhibits ALK, ROS1, and TrkA-B-C, was able to induce impressive results of responses.88–90 As ALK fusions have recently been shown to be involved in resistance to BRAF inhibitors in melanoma,91 combinatorial therapies of ALK inhibitors with other targeted therapies should be investigated in a subset of CRC patients. Much like the approval of pembrolizumab in MSI tumors, in 2019, the FDA approved the NTRK inhibitor entrectinib in NTRK-fusion-mutated tumors of all organ types, including CRC.
PI3KCA, TP53 and consensus molecular subtypes
Approximately 14–18% of CRC patients harbor mutations in the PI3KCA gene.92 A recent meta-analysis covering 12,747 patients did not demonstrate a substantial prognostic role of PIK3CA mutation status in CRC.93 On the contrary, several reports suggest that PI3KCA mutations, especially in exon 20, are linked to clinical resistance of anti-EGFR therapies.30,94 Routine testing for PIK3CA is currently not recommended. Further studies are needed to judge the role of PIK3CA as a potential predictive biomarker in CRC.
TP53 is the most frequent somatic gene mutation in all cancer types. In CRC, the mutational status of TP53 has been associated with a positive response to adjuvant 5-FU therapy.95 However, once again, more studies are necessary to determine the role of TP53 as a potential prognostic and predictive biomarker in CRC.
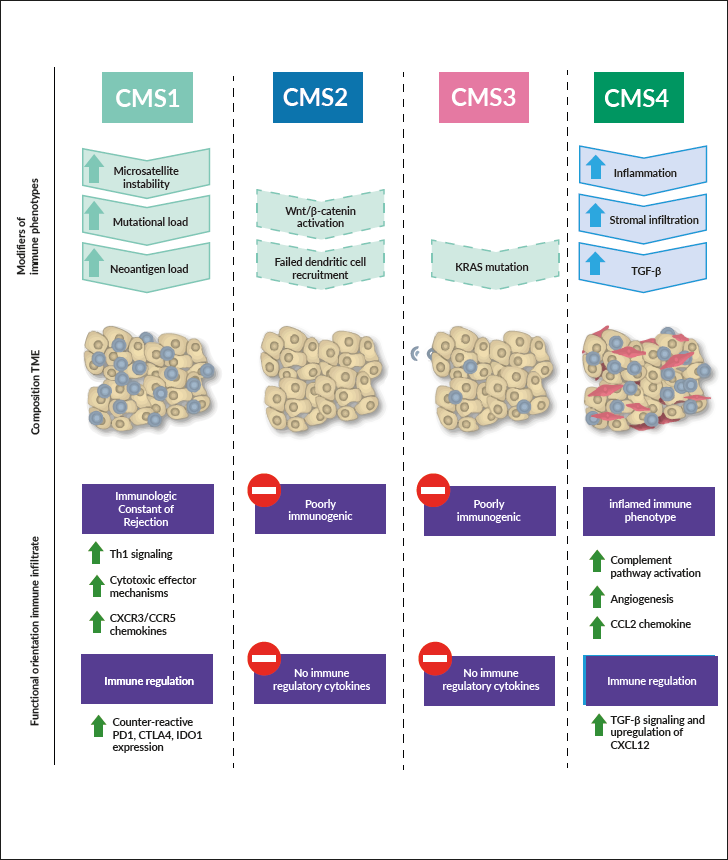
In contrast to breast cancer, the identification of various genetic subgroups of CRCs has been disappointing when applied as prognostic markers. In late 2016, four consensus molecular subtypes (CMS) based on multigene arrays and conserved across all examined studies were formed by a consortium of researchers.47 These subtypes are referred to as CMS1 (MSI-immune subgroup representing 14% of CRC cases), CMS2 (canonical subgroup accounting for 37% of cases), CMS3 (metabolic subgroup representing 13% of CRC patients), and CMS4 (mesenchymal representing 23% of CRC cases) (Figure 2). CMS subtyping primarily showed an association with clinical outcomes.96 However, even if some recent pre-clinical studies have highlighted the clinical relevance of CMS subtypes, through demonstrating differing drug efficacy between tested subtypes, the clinical impact of these subtypes remains relatively limited so far.97
_of_colorectal_cancer_and_associated_intratumoral_phenot.png)
CONCLUSIONS
Colorectal cancer is not a single disease, whereby the heterogeneity is better understood by identifying different biomarkers in the early and advanced stages. Within the evolution of targeted treatment options to these specific mutations, alterations, and fusions, the identification of such biomarkers could be used as predictive information and guidance for treatment. The field of immuno-oncology has expanded into the therapy of CRC, where subtypes such as MSI-high benefit from checkpoint inhibition with long-lasting disease control rates. However, the field of identification of these biomarkers is still young, and the knowledge of a single marker, as well the interplay between the co-occurring mutations, must be understood through further research in this field. Personalized CRC treatment is a promising step for both current and future outlooks.
TAKE-HOME-MESSAGES
-
Colorectal cancer (CRC) is a heterogeneous tumor with various predictive and prognostic biomarkers.
-
The value of testing these biomarkers in the early stage (I-III) as well-advanced stage (IV) has become a major step prior to starting a treatment or to switching to another treatment in disease history.
-
Promising upcoming biomarkers as well as the impact of liquid biopsy testing will improve and guide the therapy for CRC patients in the near future.
Conflict of Interest
AS declares no conflict of interest with this article.