
INTRODUCTION
Hepato-pancreato-biliary (HPB) cancers are almost uniformly fatal malignancies with increasing incidence and poor prognosis even when diagnosed in the early stages of the disease.1 The five-year survival rates for pancreatic ductal adenocarcinoma (PDAC) and biliary tract carcinomas (BTCs) are only 10% and 5–18%, respectively, when considering all stages, with comparably little improvement over recent years.2–5 Hepatocellular carcinoma (HCC) has a slightly better prognosis for early disease, with a five-year survival rate of 33% for localized stages, descending though to only 2% for metastatic disease.6,7 Overall, HPB cancers rank amongst the leading causes of cancer-related deaths worldwide.
For PDAC and BTCs, systemic treatment options are limited to a number of systemic chemotherapy regimens, which have shown a modest survival benefit in global tumor population-based clinical studies.8–10 Antiangiogenic agents and, more recently, immunotherapy form the basis of systemic treatment for HCC, with very significant advances over recent years in this difficult-to-treat tumor entity.11–15
A striking feature of PDAC, BTC, and HCC is the lack of predictive biomarkers to guide clinical decision making, both for multimodality curative-intent treatment and for advanced-stage metastatic disease. While molecular profiling and personalized genotype-based treatment have profoundly changed the way, we treat a wide range of tumor entities, biomarker-guided precision treatment for HPB cancers has only very recently entered the clinic, yet shows significant promise to advance the field.
Within this review, we will discuss the latest advances in precision oncology for HPB cancers, from the medical oncologist’s perspective. The most urgent goals in the clinic are to 1) personalize multimodality treatment for the localized disease to increase cure rate, 2) individualize the selection of systemic treatment for advanced disease, and 3) advance novel, genotype-specific targeted agents into the clinic through innovative clinical trial design and accelerated approval of drugs based on molecular profile.
Pancreatic ductal adenocarcinoma
Molecular pathology and genomic profiling
On the molecular level, pancreatic ductal adenocarcinomas (PDACs) are characterized by the near-ubiquitous presence of oncogenic mutations in the KRAS oncogene, found in up to 95% of tumors. Furthermore, commonly found alterations are in the tumor suppressor genes TP53, SMAD4, and CDKN2A.16 Beyond these hallmark alterations, significant inter-patient heterogeneity dominates the genetic landscape of PDAC.17 Up to 25% of PDACs harbor features of homologous recombination deficiency (HRD); precision treatment is most advanced for this sub-population.
For the molecular profiling of PDAC, we advocate a stratified testing approach. For irresectable, relapsed or metastatic disease, we test for the presence of KRAS mutation, homologous recombination deficiency (HRD), and microsatellite instability (MSI). We recommend adding further, more comprehensive testing in patients with unmutated KRAS to identify alternative driver aberrations in this molecularly and clinically distinct subgroup of PDAC.
We suggest upfront testing for HRD (or “BRCAness”) before initiation of first-line chemotherapy, to select a platinum-based first-line chemotherapy regimen for patients with HRD tumors. As there are no clear guidelines to assess HRD in pancreatic cancer, we recommend sequencing of HRD-related genes, alongside the assessment of structural chromosomal aberrations (large-scale state transitions [LST]) and loss of heterozygosity (LOH), whenever possible.18–22
Currently, we do not perform upfront BRCA1/2 germline testing, as recommended by the latest NCCN guidelines,23 due to the regulatory differences between the United States and Switzerland. However, we suggest genetic counseling for those patients with a positive family history of PDAC or other HRD-associated malignancies. This includes patients with ovarian and breast cancer, and patients whose tumors harbor HRD-related mutations, particularly when mutant allele frequency in the tumor biopsy is ≥50%.
Clinically relevant molecular subgroups
Homologous recombination deficient PDAC
DNA homology recombination repair pathway (HRR) genes are mutated in up to 25% of PDACs (Figure 1). This confers relevant therapeutic implications for both selection of chemotherapy and targeted treatment. Moreover, HRD is a very active preclinical and clinical research focus in PDAC.16

The first and, in our view, currently the most relevant consequence of HRD detected in a pancreatic tumor, is the selection of platinum agents for first-line chemotherapy. The lack of predictive biomarkers for chemotherapy in PDAC has been a long-standing issue and, given that only around 50% of metastatic PDAC patients ever receive second-line chemotherapy in a real-world setting, best-possible selection of first-line chemotherapy is crucial.24 A combined meta-analysis of 4 studies, with a systematic review of 16 studies, aimed to evaluate the impact of platinum-based chemotherapy on the prognosis of patients with resected or metastatic PDAC with HRD and showed overall survival (OS) benefit for platinum-based chemotherapy regimens for these patients.25 Also, recent randomized controlled trials selectively enrolling HRD-PDAC patients reported unexpectedly high OS for patients treated with first-line platinum agents irrespective of additional treatments.26,27 In our view, upfront testing for HRD should be performed before initiation of first-line chemotherapy, particularly when platinum-free regimens are considered. Whether HRD-PDACs should be treated with cisplatin plus gemcitabine instead of oxaliplatin combinations is an open question.26
Another consequence of HRD in PDAC is poly-ADP-ribose polymerase (PARP) inhibitor sensitivity. In recent years, PARP inhibitors (PARPis) have emerged as a potential targeted treatment option for a variety of HRD tumors, including PDAC.16 Most significantly, the phase III POLO trial showed prolonged progression-free survival (PFS) in patients with PDAC harboring BRCA1/2 germline mutations when receiving olaparib maintenance after induction platinum-based chemotherapy.27 Olaparib has since been approved in both the United States and European Union in this indication. However, the study did not report any difference in OS, and responses to PARP inhibition were profoundly heterogeneous, even within this highly selected patient population. Moreover, the therapeutic benefit of PARPis in PDACs harboring either germline or somatic HRR alterations beyond BRCA1/2 mutations is currently unknown, and no established biomarkers exist to guide personalized treatment of HRD in pancreatic cancer.28 Another maintenance trial evaluated rucaparib in patients with PDAC harboring pathogenic somatic or germline mutations in BRCA1, BRCA2, or PALB2, and without evidence of progression on previous platinum-based chemotherapy in a phase II setting, demonstrated a promising disease control rate of 89.5%.29 Furthermore, a recent phase II trial investigating the combination of veliparib, a PARPi, with first-line chemotherapy (cisplatin and gemcitabine), showed no additional benefit but increased toxicity in advanced PDACs with germline BRCA1/2 or PALB2 mutations.26
An exciting field of clinical and translational research is the combination of PARPis with other therapeutic agents. An ongoing randomized phase II trial is evaluating the combination of niraparib plus nivolumab or ipilimumab in patients with advanced PDAC without tumor progression for ≥16 weeks on platinum-based therapy (NCT03404960).30 Additionally, synergistic combinations of PARPis with further targeted agents (such as cell cycle, bromodomain and extra terminal domain [BET], or mitogen-activated protein kinase [MAPK] pathway inhibitors) are under preclinical investigation in order to optimize responses.31–33
In summary, PARP inhibitor treatment, either as a single drug or in combination, is a promising therapeutic strategy in PDAC patients with HRD. However, no studies so far have shown benefit in OS, and biomarkers predictive of response have still to be developed.
KRAS wild type PDAC
Five-to-eight percent of PDACs do not harbor a KRAS driver mutation. This clinically and molecularly distinct subgroup of PDAC has moved into the focus in recent years after several reports described successful therapeutic targeting of alternative oncogenic driver alterations found in this subgroup. KRAS-wild type (WT) PDAC driver alterations include oncogenic fusions, amplifications, and oncogenic mutations. Recurrent NRG1 rearrangements have been reported in KRAS-WT PDACs and are therapeutically targetable with ERBB inhibitors, such as afatinib or erlotinib-pertuzumab combination therapy.34 Less frequently, RET fusions can also drive PDAC and are amenable to treatment with tyrosine kinase inhibitors (TKIs).34 BRAF alterations are found in about 3% of PDACs, including the V600E mutation, targetable with combined BRAF and MEK inhibition.35,36 Additionally, approximately 1% of the KRAS-WT PDACs harbor class 2 or class 3 BRAF alterations, including in-frame insertions or deletions, such as BRAF p.N486_P490del or p.T599dup.37 These lack response to class 1 BRAF inhibitors but might respond to MEK inhibition.37,38
For the clinic, it is essential that all PDACs undergo KRAS testing, primarily to identify KRAS-WT tumors. These tumors should then undergo more comprehensive molecular profiling to uncover alternative oncogenic drivers in this subgroup.
Mismatch repair-deficient/high microsatellite instable PDAC
PDAC’s microenvironment is highly immunosuppressive39–41 leading to marginal immunotherapy activity.42–44 The only exceptions are the 1–2% of PDACs exhibiting mismatch repair deficiency/high microsatellite instability (dMMR/MSI-H). Similar to other dMMR/MSI-H tumors, dMMR/MSI-H PDAC showed some profound and durable responses to checkpoint inhibition in clinical trials.45,46
Currently, the programmed cell death-1 (PD-1) inhibitor pembrolizumab is Food and Drug Agency (FDA) approved for second-line treatment of solid tumors with dMMR/MSI-H, including PDAC.47,48 In our opinion, all PDACs should undergo testing for mismatch repair deficiency, even if the pre-test probability is very low.
Novel therapeutic agents
Single nucleotide variant-selective direct KRASG12C inhibitors represent the first-ever KRAS inhibitors with clinical activity.49,50 Approximately 3% of PDACs harbor KRAS G12C mutations.51 Amongst KRASG12C inhibitors, AMG 510 (sotorasib) is currently leading the field, with very promising response rates as monotherapy in non-small cell lung carcinoma (NSCLC).52 Preliminary data from the phase I trial in colorectal cancer and other solid tumors harboring KRAS G12C, showed stable disease in six of the eight patients with refractory PDAC, with three patients achieving a 30% decrease of tumor burden. Most strikingly, AMG510 is well-tolerated with significant potential for combination treatment.
Other novel molecularly targeted agents and immunotherapeutics have shown more disappointing results. The combination of nivolumab and the colony-stimulating factor 1 receptor (CSF1R) antibody, cabiralizumab recently failed to improve PFS in advanced microsatellite stable (MSS)-PDAC.53 Furthermore, pegylated recombinant human hyaluronidase (PEGPH20), in combination with gemcitabine and nab-paclitaxel, failed to demonstrate any benefit in PDAC with high-levels of hyaluronic acid in the phase III trial HALO-109-301, leading to an additional increase in thromboembolic events.54,55
Major efforts in the field are dedicated to the development of novel prognostic and predictive biomarkers based on comprehensive molecular profiling. The COMPASS trial aimed to uncover predictive signatures for targeted treatment selection by performing whole genome sequencing and RNA sequencing in advanced PDAC. Potentially targetable alterations could be detected in 30% of the analyzed samples, and gene expression signatures of both tumor cells and tumor stroma are being developed for clinical application.56
Additionally, major efforts are being done to optimize patients’ monitoring and individualize therapeutic approaches. PDAC is characterized by a highly aggressive course of the disease, leading to rapid resistance to systemic treatment and high rates of relapse, even after a curative-intentioned approach for localized disease. Serial liquid biopsy with longitudinal monitoring of circulating mutant KRAS is emerging as a promising tool towards enabling personalization of patients’ follow-up.57,58
Biliary tract carcinoma
Molecular diagnostics
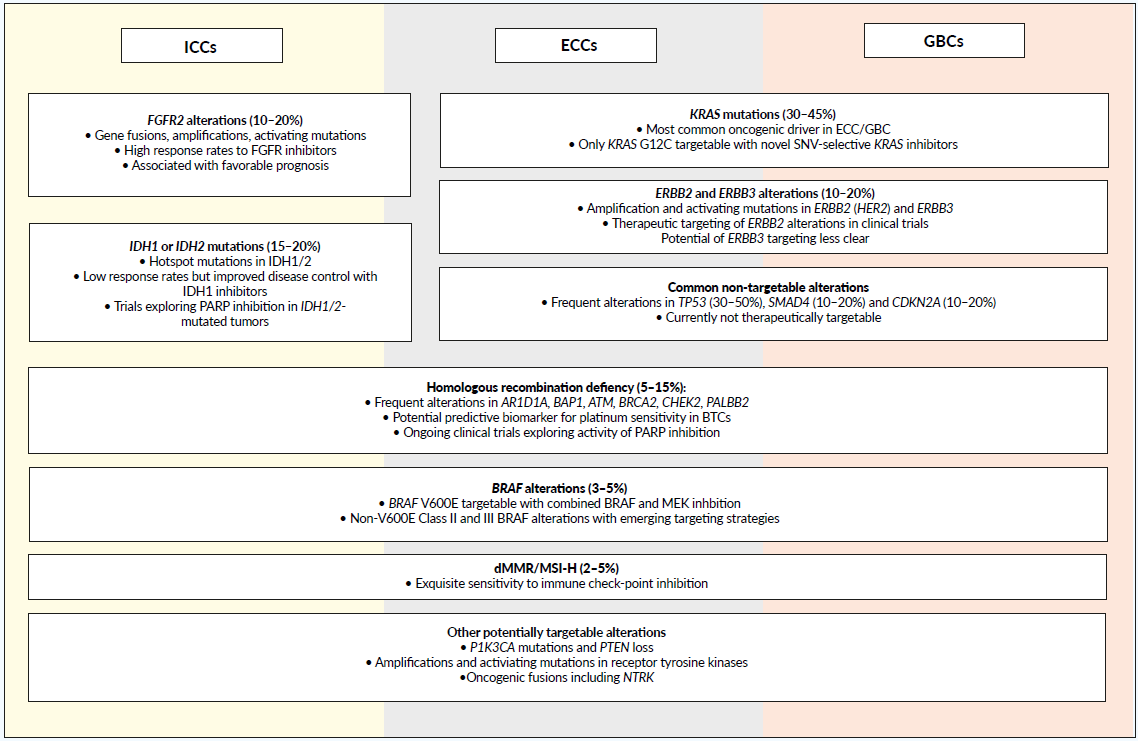
Biliary tract carcinomas (BTCs) are a clinically and genetically heterogeneous group of malignancies arising from the biliary epithelium. While all four anatomical subgroups of BTCs (intrahepatic, ICC; perihilar, PCC; distal extrahepatic, ECC; gallbladder, GBC) in principle show overlapping genetic features, the prevalence of individual molecular subgroups varies significantly between these groups (Figure 2). ICCs show the largest proportion of molecular subgroups with targetable genetic alterations (up to 50%), while hitherto undruggable driver alterations are more common in PCC/ECC and GBC. Still, in our view, all irresectable or metastatic BTCs should undergo comprehensive molecular profiling as early as possible. Currently, the most relevant therapeutically targetable molecular alterations in BTC are fibroblast growth factor receptor 2 (FGFR2) alterations and isocitrate dehydrogenase 1/2 (IDH1/2) mutations, both commonly found in ICC. Beyond that, several smaller subgroups, including those harboring BRAF V600E, ERBB2 amplifications of mismatch repair deficiency, are currently clinically relevant. Extrahepatic cholangiocarcinomas frequently exhibit not-yet-targetable alterations, such as KRAS (30–45%) and TP53 (30–50%) mutations.59 Gallbladder cancers harbor ERBB2/3 amplifications and CDKN2A/B loss relatively frequently and are, therefore, potential molecular targets.60
._prevalence_of_targetable_.png)
Particularly for locally advanced irresectable non-metastatic ICCs, molecular testing should be performed upfront, given that FGFR inhibition in FGFR2 altered tumors and, more rarely, combined BRAF and MEK inhibition for BRAF V600E mutated tumors, shows excellent objective response rates (ORR), superior to those achieved with chemotherapy in historical trials, suggesting tremendous potential for conversion treatment.
Molecular testing should cover the most commonly reported oncogenic mutations and fusions, as well as DNA copy number alterations. More experimental is still the assessment of structural chromosomal aberrations associated with HRD (“BRCAness”), including LST and LOH.
Molecular subgroups with clinical relevance
BTCs with FGFR alterations
Up to 20% of ICCs harbor oncogenic FGFR alterations, including oncogenic fusions and rearrangements, activating mutations or gene amplifications.61,62 Aberrations in FGFR signaling promote tumor cells survival, proliferation, and invasion.63 In BTCs, the most common FGFR alterations are FGFR2 fusions.61,64 This is particularly relevant since patients with tumors exhibiting FGFR2 fusions have shown response rates under selective FGFR inhibitors such as erdafitinib and pemigatinib.65–68 Proven high response rates also make these agents particularly attractive for conversion treatment of locally advanced tumors. Erdafitinib is currently FDA approved for metastatic urothelial carcinoma after progression on platinum-based chemotherapy. However, early access programs are available for other tumors, including BTCs.
Pemigatinib, a FGFR 1-3 inhibitor, showed a response rate of 35.5% in a recently published phase II trial (FIGHT-202), including pretreated advanced cholangiocarcinomas (CCs) with FGFR2 fusions.69 Based on this trial, pemigatinib gained FDA approval for BTCs with FGFR2 fusions and rearrangement. Both pemigatinib and infigratinib, another selective FGFR 1-3 inhibitor, are currently being compared to first-line chemotherapy with cisplatin and gemcitabine in a phase III trial for CCs with FGFR2 fusions or translocations (NCT03656536, NCT03773302).70
Further, acquired resistance mechanisms to FGFR inhibition are being investigated, and reports of drug combinations aiming to overcome this resistance, such as synergistic combinations with mammalian target of rapamycin (mTOR) inhibitors, have recently been published.65 Taken together, FGFR inhibition in BTCs harboring oncogenic FGFR alterations hold significant therapeutic potential and undergo rapid clinical development.
IDH1/2 mutated BTCs
IDH1/2 alterations are present in up to 20% of ICCs.71–74 IDH1/2 hotspot mutations are neomorphic, leading to accumulation of D-2-hydroxyglutarate, an oncogenic metabolite causing epigenetic changes and cellular dedifferentiation.75 Recently, the IDH1 inhibitor ivosidenib showed significant improvement in PFS in a phase III study as compared to placebo in pretreated IDH1-mutated advanced BTCs, and has been recently FDA approved for this indication.76,77 The results of phase I/II trial of enasidenib (AG-221), an IDH2 inhibitor, in relapsed IDH2 mutated solid tumors (NCT02273739) was completed in 2016, still pending publication.78 Characteristically, disease stabilization is the predominant outcome of treatment with IDH inhibitor in ICCs, with very low objective response rates. Ongoing research is therefore exploring combination strategies.
Homologous recombination deficient BTCs
Five-to-fifteen percent of BTCs harbor defects in the HRR pathway, with the most frequent alterations located in ARID1A, BAP1, ATM, BRCA2, CHEK2, and PALB2 genes.19 Additionally, functional defective double-strand DNA repair has been associated with the presence of IHD1/2 mutations, suggesting potential vulnerability to PARPis.79,80 A phase II trial with olaparib for advanced refractory IHD1/2 mutated solid tumors, including BTCs, is currently recruiting (NCT03212274). Moreover, several trials study the potential of PARP inhibition in HRD-BTCs.
Other molecular subgroups
Roughly 2–4% of ICCs, and about 5% of GBCs and ECCs, are dMMR/MSI-H and, thus, respond to immunotherapy.81 Additionally, a further 3–5% harbor the BRAF V600E mutation, leading to high objective response rates to synergistic BRAF and MEK inhibition.82 Targeting strategies for ERBB2-amplified BTCs are emerging83 while the role of HER2 and HER3 mutations as therapeutic targets are currently less clear.
Novel therapeutic agents
The combination of the antiangiogenic drug lenvatinib with the anti-PD1 checkpoint inhibitor pembrolizumab showed promising activity in early phase trials84 and is in further clinical development for BTCs (NCT03895970).
Hepatocellular carcinoma
Molecular diagnostics
The clinical diagnosis of hepatocellular carcinoma (HCC) is based on radiologic criteria, and historically biopsies have only been taken in unclear situations. Molecular analyses of HCCs are still not a clinical standard, and there is debate as to whether a proportion of HCC patients could benefit from comprehensive molecular profiling and personalized genotype-based treatment. This is also due to the fact that the most frequently found genomic alterations in HCC are currently non-targetable, such as TERT (47%) or TP53 (29%).85
However, independent of molecular profiling, many new therapeutic options for advanced HCC have emerged in recent years, increasing tumor response rates, and patients´ survival.14,15 These recent advances entail an urgent demand for the development of novel biomarkers able to predict response and personalize treatment choice. In summary, the current clinical translation of molecular profiling in HCC remains an unresolved challenge.
Emerging molecular targets and biomarkers with potential clinical relevance
Among molecular alterations found in HCC, FGF19 amplifications associated with FGFR4 pathway hyperactivation show some potential as a therapeutic target. The FGF19/FGFR4 axis has been shown to be a relevant driver of carcinogenesis in HCC, providing new insights into its molecular background and uncovering novel therapeutic vulnerabilities of these tumors.86 The first FGFR4 selective inhibitors (fisogatinib) are currently in early phases of clinical development for HCC treatment.87
Establishing predictive biomarkers for the rapidly emerging systemic treatment options for advanced and metastatic HCC is a major goal of HCC precision oncology. Efforts have been made to analyze differential responses in clinical subgroups of patients included in trials. In the phase III trial of the first-line atezolizumab combined with bevacizumab, randomized against sorafenib, patients with viral etiology of HCC seemed to have a higher benefit from the combination.15 On the contrary, patients with non-viral etiology showed better responses in the first-line lenvatinib phase III trial.14 Based on preliminary data, only patients with α-fetoprotein concentrations of 400 ng/mL or higher were eligible for the phase III trial of ramucirumab after first-line sorafenib.88
Role of immunotherapy
Recently immunotherapy has arisen as a promising systemic treatment option for patients with HCC. However, biomarkers predicting response to checkpoint inhibitors, such as PD-L1 status, have failed to correlate with tumor responses.14,89–91
Results from the CheckMate-040 study lead to the recent FDA approval of ipilimumab and nivolumab for advanced HCC, progressing after the previous sorafenib.92
For first-line treatment, atezolizumab and bevacizumab have shown in a phase III trial improvement in OS and PFS, as compared to sorafenib, and have received FDA approval for this indication.15 The combination of lenvatinib and pembrolizumab has shown a response rate of 36% with a disease control rate of 88% in a first-line phase Ib study for HCC not amenable for locoregional treatment.93 A phase II trial in the first-line setting is ongoing (NCT03895970).
FUTURE DIRECTIONS OF PRECISION ONCOLOGY OF HPB CANCERS
Precision oncology is rapidly changing the way we approach the diagnostics and treatment of HPB cancers. With the recent approval of molecularly targeted agents for PDAC, BTC, and HCC, precision treatment has entered routine clinical management of HPB cancers, bringing long-awaited progress but also significant challenges. In our opinion, early and stratified molecular characterization of HPB tumors is a fundamental requirement for the successful implementation of precision oncology for HPB tumors. A relevant challenge is to integrate molecular profiling in earlier disease stages, which requires interdisciplinary workflow and assessment of more patients through a molecular tumor board. Furthermore, the time-point of molecular profiling, as well as a need for re-biopsies over disease course due to adaptive tumor clonal evolution under treatment, are important questions to be addressed.
On the other hand, evaluation of smaller molecularly categorized subgroups of patients in clinical studies would require novel, more flexible trial designs, allowing rapid translation of research data into the clinic. Additional efforts are also needed to develop standardized biomarkers to predict response and resistance to treatment. In this sense, liquid biopsy is a promising tool permitting non-invasive longitudinal monitoring of disease. A further relevant challenge for establishing precision oncology in the clinic is to develop rational algorithms of molecular profiling to keep it cost-efficient and sustainable. Conversely, increased rates of molecular profiling require additional optimization of germline testing and counseling.
CONCLUSION
Precision oncology of hepato-pancreato-biliary (HPB) cancers is a rapidly developing field, enabling personalized clinical management of patients, therefore, offering them more effective and broader therapeutic opportunities. Nevertheless, it remains a significant challenge to optimally integrate the advances made in basic and translational research into the clinic. Therefore, the establishment of rationale diagnostic algorithms and innovative designs of clinical trials are urgently needed to validate preclinical data and enable early access to novel drugs.
In summary, we believe precision oncology has fantastic potential to transform the management of HPB cancer patients, but still relevant efforts are needed to integrate this highly dynamic and promising field into the daily clinical practice.
TAKE-HOME MESSAGES
-
Precision oncology of hepato-pancreato-biliary (HPB) cancers is a rapidly developing field, with great potential to individualize and improve clinical management of patients.
-
BRCA-mutated pancreatic cancers, biliary tract carcinomas harboring FGFR2 or IDH1 alterations, and tumors with mismatch repair deficiency/high microsatellite instability or high tumor mutational burden (TMB) are subentities with already FDA-approved precision treatment options.
-
More rapid integration of precision oncology for HPB cancers into clinical practice requires more standardized diagnostic algorithms and innovative trial designs.
-
Molecular monitoring and more complex integrated biomarkers hold great potential to personalize multimodality treatment of HPB cancers in the future.
CONFLICT OF INTEREST
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
All authors contributed to and approved the final manuscript.