INTRODUCTION
Gastro-esophageal adenocarcinomas (GEA) are a leading cause of cancer mortality, with gastric cancer alone representing the sixth malignant neoplasm by incidence (11.1 per 100,000/ year) and the fifth cause of cancer mortality worldwide (8.2 per 100,000/year) according to GLOBOCAN data.1 They represent a highly heterogeneous group of tumors, and it is expected that the identification of well-defined subtypes–each with their own specificities in terms of oncogenetic drivers, survival strategies, and drug susceptibility–might be helpful in defining more efficient treatment strategies. This paradigm that can be collectively defined as “precision oncology,” unfolds through the historical approach of targeted treatment and the more recent approach of immunotherapy. Both strategies rely on the identification of accurate biomarkers to attribute the correct drug to the right patient.
CURRENT CLASSIFICATIONS
The first demonstration that genetic signatures could be prognostic, and possibly predictive of treatment response in GEA, came from the classification proposed by Tan et al. (2011), dividing gastric cancers into two subtypes: genomic intestinal (G-INT) and genomic diffuse (G-DIF).2 This categorization, while it did not show correspondence with the historical Lauren classification, identified G-DIF as a subtype with worse prognosis, and suggested differential sensitivity to chemotherapeutics in vitro, with G-INT more sensitive to 5-fluorouracil (5-FU) and oxaliplatin and G-DIF more sensitive to cisplatin. Moreover, G-INT appeared to be associated with more significant clinical benefit from 5-FU-based adjuvant therapy.
Efforts from The Cancer Genome Atlas (TCGA) collaboration have led to another proposed classification, dividing GEA into four main molecular subtypes, namely: Epstein-Barr virus (EBV) positive cancers, tumors with microsatellite instability (MSI), with chromosomal instability (CIN), and genomically stable (GS) cancers (Figure 1).3 Each subtype shows specific genetic and epigenetic alterations that may pave the way to targeted clinical trials.
-
EBV-associated cancers show a high prevalence of DNA hypermethylation (e.g., CpG island methylator phenotype (CIMP)) and phosphatidylinositol 3-kinase, catalytic subunit alpha (PIK3CA) mutation. PIK3CA mutation is present in up to 80% of EBV-associated GEA, in contrast with a prevalence of 3–42% in other subtypes. Moreover, expression of immune checkpoint proteins such as programmed death-ligand 1/2 (PD-L1/2) is elevated, suggesting that this subtype may benefit from the treatment with checkpoint inhibitors.
-
The MSI subtype is characterized by PD-L1 expression and frequent occurrence of potentially targetable mutations in PIK3CA, ERBB3, ERBB2, and epidermal growth factor receptor (EGFR). Furthermore, the defining alteration in the MSI subgroup opens up a clinically available treatment option with the immune checkpoint inhibitor pembrolizumab.
-
The CIN subtype shows genomic amplification of receptor tyrosine kinases (RTKs). Angiogenesis is hypothesized to have a significant role in this tumor subtype, due to frequent detection of elevated vascular endothelial growth factor A (VEGF-A).
-
GS gastric cancers are characterized by mutations in cell adhesion- and angiogenesis-related pathways, such as E-cadherin, the RAS homolog gene family, member A (RHOA) gene, or fusions involving Rho-family GTPase-activating proteins (GAPs). The GS subtype appears to be enriched for the diffuse histological variant.2
The Asian Cancer Research Group (ACRG)-proposed classification for GEA identifies four subtypes: MSI-high, microsatellite stable/epithelial-mesenchymal transition (MSS/ EMT), microsatellite stable/epithelial/TP53 intact (MSS/ TP53+, p53 active), and microsatellite stable/epithelial/TP53 loss (MSS/TP53-, p53 inactive).4
-
MSI-high cancers were prognostically more favorable, had lower recurrence rates, and often showed hypermutation in genes such as ARID1A (44.2%), the PI3K-PTEN-mTOR pathway (42%), KRAS (23.3%), and ALK (16.3%). Histology was predominantly intestinal.
-
MSS/EMT tumors were characterized by a younger age at diagnosis, high prevalence of diffuse histology, and molecular alterations in pathways related to cell adhesion (such as E-cadherin), motility, and angiogenesis. They were prognostically unfavorable with a high recurrence rate.
-
The MSS/TP53+ subtype had the highest prevalence of TP53 and RHOA mutations, with a predominance of intestinal histology and male sex; it also presented mutations in APC, ARID1A, KRAS, PIK3CA, SMAD4. This subgroup had the highest prevalence of EBV infection.
-
MSS/TP53- tumors had the second-best prognosis, showed TP53 mutations in up to 60% of cases, and had a low frequency of other mutations, although recurrent focal amplifications in ERBB2, EGFR, CCNE1, CCND1, MDM2, ROBO2, GATA6, and MYC were frequent.
The reported data seem to suggest a partial overlap between the TCGA and ACRG classifications; specifically, one could suggest a correspondence between the EBV, MSI, CIN, and GS subtypes and the MSS/TP53+, MSI, MSS/TP53-, and MSS/EMT subtypes, respectively. However, the CIN and GS subtypes can be identified across all four ACRG groups; and, when closely comparing the GS and MSS/EMT subtypes, it can be observed that the frequency of E-cadherin mutations is dissimilar, and RHOA mutations, frequent in GS cancers, are conversely rare in MSS/EMT and more prevalent in MSS/ TP53+ and MSS/TP53-. Multiple reasons can be adduced to justify these discrepancies, from variability in patient populations to tumor sampling and analysis techniques. Of note, multiple authors have attempted to employ less expensive and more widely available techniques such as immunohistochemistry and in situ hybridization to mirror these classifications.
Understandably, a possible limitation of these classifications lies in the fact that the analyses are usually carried out on samples from a single primitive or metastatic site, therefore, they cannot reflect tumor heterogeneity and clonal evolution. As evidenced by Pectasides et al. (2018), mutational discordance between the primary tumor and secondary lesions may be as frequent as 40–60%.5 Analysis of circulating tumor DNA (ctDNA) has been proposed as a strategy to circumvent these limitations; however, the authors show that not all genetic alterations present in the primary tumor can be detected on ctDNA, and vice versa. It also remains to be determined whether treating patients based on ctDNA findings will impact outcomes.
Recently, epigenetic signatures have been investigated as possibly predictive of therapeutic benefit. Sundar et al. (2019) studied DNA methylation on samples from the MRC OE02 trial, which randomized patients with resectable esophageal cancer to surgery alone or neoadjuvant chemotherapy followed by surgery.6 An epigenetic methylation signature was identified to be predictive of chemotherapy benefit.
CURRENT STRATEGIES AND FUTURE DEVELOPMENT: WHERE ARE WE CURRENT CLASSIFICATIONS
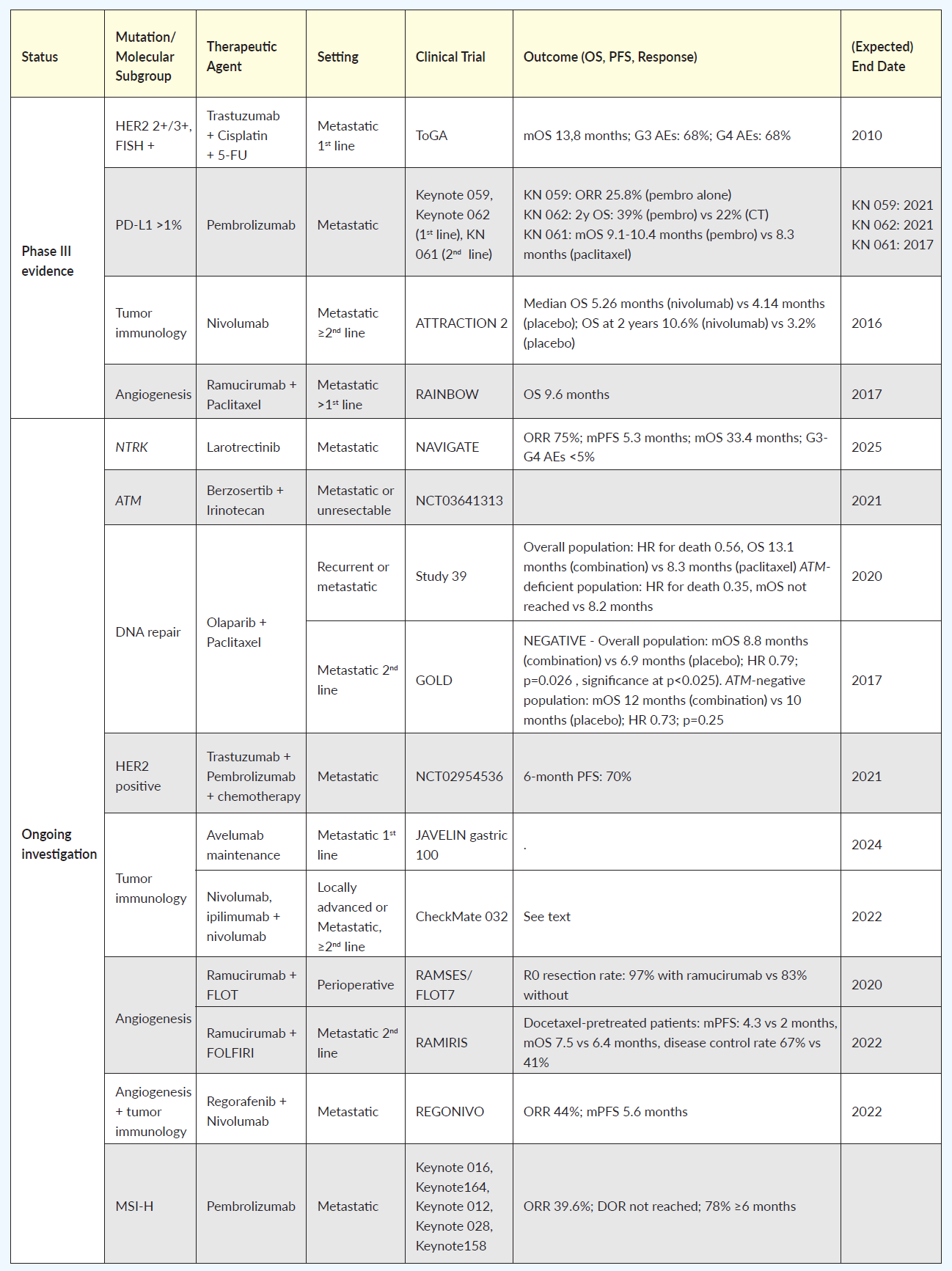
Table 1 summarizes the most relevant clinical trials, completed or ongoing, classified by a specific molecular target. Currently, c-ERB-B2 (HER2) is one of the few biomarkers with a recognized therapeutic role in common clinical practice in GEA, based on results from the ToGA trial showing overall survival (OS) benefit for HER2 overexpressing tumors by immunohistochemistry or gene amplification by fluorescence in situ hybridization, in the first-line setting, when treated with trastuzumab in addition to cisplatin and a fluoropyrimidine (5-FU or capecitabine) chemotherapy.7 The JACOB phase III trial was an attempt to build on these results by the addition of pertuzumab to the schedule, in a dual HER2 blockade.8 Although a 3.3-month increase in OS was achieved, the study failed to demonstrate a statistically significant advantage. The reason may have something to do with gastric cancer’s heterogeneity and weak membrane staining, as compared with breast cancer (where the dual blockade is superior to single). The PETRARCA study, recently presented at the American Society of Clinical Oncology (ASCO) meeting, investigated the use of the same anti-HER2 agent combination in the perioperative setting, showing improved pathologic complete response rates and lymph-node negativity rates at the cost of more toxicity; also due to the negative results of JACOB, this trial was closed prematurely and did not proceed to phase III.9 Microsatellite instability (MSI) is another biomarker with a recognized therapeutic implication. In 2017, the immune checkpoint inhibitor pembrolizumab was granted accelerated approval by The Food and Drug Administration (FDA) for the treatment of patients with unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment option.10 This indication was therefore approved without regard to the primary site, in a so-called “tumor-agnostic approach.”
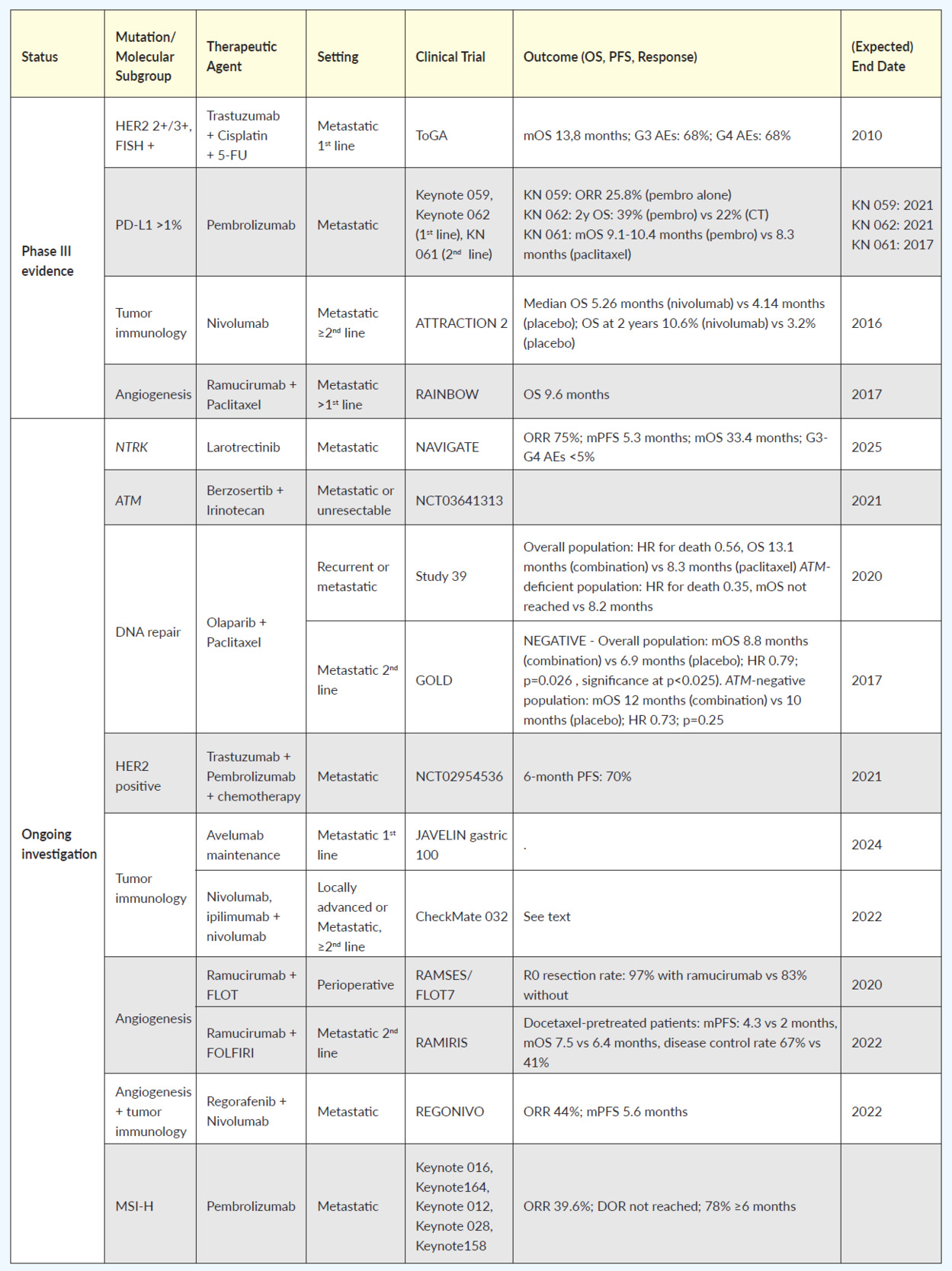
Focusing on GEA, in the Keynote 059 trial, pembrolizumab was tested on a population of patients who had previously received at least two lines of treatment; objective response rate (ORR) was 15.5% and 6.4% for PD-L1 positive and PD- L1 negative tumors, respectively, and the median duration of response was 16.3 and 6.9 months.11 The role of PD-L1 as a potential biomarker was further investigated in the Keynote 062 study that tested pembrolizumab versus chemotherapy in the first-line setting of GEA, with PD-L1 expression >1%. Results confirmed the non-inferiority of pembrolizumab in the overall study population and suggested a greater benefit for patients with PD-L1 expression >10% in a subgroup analysis.12
A further experience by Kim et al. (2018) on 61 patients with metastatic gastric cancer, has shown that exceptional response rates of 85.7% and 100% can be obtained with pembrolizumab, in a second-line setting, in the MSI and EBV TCGA subgroups, respectively.13 Interestingly, PD-L1 positivity alone identified a population with 50% ORR, and no PD-L1 negative patients responded.
Long-term results from the phase III Keynote 061 trial, focusing on pembrolizumab versus paclitaxel in the second-line setting in PD-L1 positive patients (CPS>1), were recently made available at the ASCO 2020 annual meeting.14 The study showed an improvement in OS and duration of response with pembrolizumab in the CPS>1 population, and an improvement in ORR in the CPS>10 subgroup, with fewer adverse events (53% vs 84%).
Interestingly, an exploratory analysis demonstrated a strong association between a high tumor mutational burden (TMB, defined as >175 mutations per exome) and response to pembrolizumab in terms of ORR, progression-free survival (PFS) and OS.15 Patients with high TMB reached a median OS of 16.4 months if treated with pembrolizumab and 8.1 months if treated with paclitaxel. On the other hand, patients with low TMB performed worse when treated with pembrolizumab (median OS 5.7 months) rather than paclitaxel (median OS 8.8 months). However, these data are exploratory, and TMB does not currently have a role in the therapeutic decision on the use of checkpoint inhibitors.
Of note, tumor PD-L1 status did not appear to be predictive of survival in the phase III ATTRACTION 2 trial. This trial evaluated the efficacy of nivolumab monotherapy versus placebo in chemotherapy-refractory gastric and gastro-esophageal junction cancers in an Asian patient population.16
Pietrantonio et al. (2019) evaluated the role of MSI status in a patient-level meta-analysis of the CLASSIC, MAGIC, ARTIST, and ITACA-S trials focused on patients with resectable gastric cancer.17 Besides the prognostic significance of this biomarker, as shown by better outcomes in MSI-high rather than MSI-low cancers, patients with MSI-high gastric cancer did not appear to benefit from the addition of chemotherapy to surgery. Specifically, 5-year OS with surgery plus chemotherapy versus surgery alone was 62% vs 53%, respectively, in MSI-low cancers and 75% vs 83% respectively in MSI-high cancers. The authors suggest that chemotherapy omission and immune checkpoint inhibitor therapy in this setting should be investigated prospectively. Nowadays, evidence from clinical trials supports the use of ICIs in advanced and chemo-refractory treatment settings in patients with PD-L1 positive adenocarcinoma. Nonetheless, MSI, as well as EBV, can be used as effective predictive biomarkers and should always be tested upfront.
The tumor-agnostic approach has also been proposed in the setting of neurotrophic tropomyosin receptor kinase (NTRK) gene fusions; the NTRK inhibitor larotrectinib has received FDA approval for all solid cancers harboring NTRK gene fusions.18 These appear to be infrequent in GEA and are not usually tested for in clinical practice; however, it is possible that wider use ofextensive molecular profiling may bring to more frequent identification of these rare cases. To our knowledge, no specific data are yet available on the efficacy of NTRK inhibitors in GEA; however, the efficacy and safety of larotrectinib in gastrointestinal cancers has been investigated in a recent phase II trial.19
Strategies that target angiogenesis have been explored in GEA, with the most intriguing results stemming from the combination of ramucirumab and paclitaxel in the RAINBOW trial. Here, a significant survival benefit was observed in comparison to chemotherapy alone; however, no predictive biomarkers were identified.20 Previously, the addition of bevacizumab to standard chemotherapy in the AVAGAST study had failed to improve outcomes, although neuropilin-1 expression and pVEGF-A levels emerged as possibly predictive of benefit in a subgroup analysis.21
In addition, the phase II RAMIRIS trial evaluated the efficacy and safety of a combination of FOLFIRI and ramucirumab versus paclitaxel plus ramucirumab in the second-line setting.22 Results were presented as a poster at the ASCO 2020 annual meeting. Although no significant differences in OS or PFS were detected in the intention-to-treat population, patients previously exposed to docetaxel appeared to benefit from the FOLFIRI plus ramucirumab combination (mPFS: 4.3 vs 2 months, mOS 7.5 vs 6.4 months, disease control rate 67% vs 41%); a phase III trial is ongoing. In the perioperative setting, results from the phase II RAMSES/FLOT7 study, that tested the addition of ramucirumab to perioperative FLOT, have recently been made available and show an improvement in R0 resection rate (97% vs 83%); however, a trend towards higher mortality in patients with type I gastro-esophageal junction cancer treated with ramucirumab was observed.23
The phase Ib REGONIVO trial tested low-dose regorafenib plus nivolumab in 50 patients, 25 of which had a diagnosis of gastric cancer without microsatellite instability; all patients had received at least 2 previous lines of treatment, including anti-angiogenetic inhibitors in 96% of patients, and 7 patients with gastric cancer had been treated with immune checkpoint inhibitors.24 The rationale for the combination of regorafenib plus an anti-PD-1 is the superior tumor growth suppression compared to either treatment alone exhibited in murine models. This intriguing benefit may be due to the reduction of tumor-associated macrophages by regorafenib, increasing the tumor’s sensitivity to nivolumab. The results were encouraging, with ORR of 44% and a median PFS of 5.6 months. This could be the first step towards a successful combination strategy targeting both angiogenesis and tumor immunology. It would be interesting to understand whether responders fall into any of the subtypes identified by current classifications.
Recently, Janjigian et al. (2019) tested three different immunotherapy schedules (with nivolumab 3 mg/kg alone, nivolumab 3 mg/kg + ipilimumab 1 mg/kg, and nivolumab 2 mg/kg + ipilimumab 3 mg/kg) in a second and further- line setting in the phase I/II CheckMate 032 study.25 ORR was 12%, 24% and 8%, respectively. 1-year PFS rates were 8%, 17%, and 10%, respectively; 1-year OS rates were 39%, 35%, and 24%. Treatment-related grade 3/4 adverse events were reported in 17%, 47%, and 27% of patients, respectively. Responses were observed regardless of PD-L1 status and microsatellite instability; however, PD-L1 expression and an MSI-H profile appeared to influence ORR. The same author also assessed the safety and efficacy of first-line pembrolizumab and trastuzumab plus chemotherapy in HER2-positive GEA in a single-arm phase II trial. Here, a 70% 6-month PFS was observed, and PD-L1 expression did not appear to correlate with clinical benefit;26 a phase 3 trial is underway.
The fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) axis has been identified as a contributor to gastric tumorigenesis and a possible therapeutic target. For instance, FGFR2 amplification appears to be related to worse OS, where its expression has a prognostic role in adjuvant chemotherapy failure.27,28 Several potential therapeutic agents targeted at this axis, in the form of anti-FGFR monoclonal antibodies, FGF traps, FGFR selective or non-selective inhibitors, have been developed and are being investigated. However, clinical data are still scarce.29,30
Strategies aimed at affecting the DNA damage response capabilities of cancer cells are also being explored. The ataxia-telangiectasia mutated gene (ATM) is mutated in approximately 10% of gastric cancers, and ataxia- telangiectasia and RAD3-related 1 (ATR-1) inhibition displays synthetic lethality in ATM-deficient tumors and is being explored in early phase I trials as a potential therapeutic strategy.31 PARP inhibitors are also being evaluated in this setting. A phase II study by Bang et al. (2015) tested a combination of olaparib plus paclitaxel versus paclitaxel alone in patients with recurrent or metastatic GEA, and showed a significant OS improvement in the study population (HR: 0.56, OS 13.1 months with the combination vs 8.3 months with paclitaxel alone); the benefit appeared to be greater for the ATM-deficient population (HR: 0.35, median OS not reached vs 8.2 months, respectively).32 A subsequent phase III trial (GOLD) testing the same combination in a second-line setting was formally negative, failing to show a benefit in OS between the treatment groups in either the overall or ATM-deficient population. Nonetheless, it should be noted that only 19% of patients were ATM-deficient, as opposed to 51% in phase II study.33
Given the acceptable safety of the combination of olaparib with chemotherapy, further studies with PARP inhibitors are ongoing: a phase I/II trial of olaparib in combination with ramucirumab, and another phase I trial investigating veliparib plus FOLFIRI in advanced solid tumors including GEA.34,35 The TP53 signaling pathway represents another attractive target. Efforts are being made to identify strategies to either restore the function of wild-type TP53 or pinpoint TP53-dependent targets that may be more easily addressed. There are no clinical data concerning the earlier approach in GEA; as for the latter strategy, the WEE1 protein kinase, involved in the G2-M cell cycle checkpoint, has been identified as a possible therapeutic target. Thus, a phase II trial is investigating a WEE1 inhibitor in combination with paclitaxel, in the second-line setting, in TP53 expressing gastric cancer.36 Inhibition of the TP53/MDM2 interaction, which leads to TP53 degradation, is also being addressed as a potential therapeutic strategy.37
Increased expression of MET has been observed in intestinal-type gastric cancer and appears to possess a negative prognostic value.38 Targeting of the hepatocyte growth factor/MET (HGF/MET) axis is potentially interesting and is being investigated; a randomized phase II trial evaluated ECX chemotherapy plus either the HGF-directed monoclonal antibody rilotumumab or placebo in the first-line setting, demonstrating an OS benefit (10.6 vs 5.7 months in MET positive patients).39 The subsequent phase III RILOMET-1 study, however, did not confirm this benefit, and the RILOMET-2 trial had to be closed for a safety review.40 Similarly, onartuzumab, another anti-MET monoclonal antibody, yielded unsatisfactory results in combination with FOLFOX in the RILOMET-2 trial.41
The mTOR/PI3K/AKT signaling pathway has also been proposed as a potential therapeutic target; however, the efficacy of this approach has not yet been demonstrated, with everolimus performing unsatisfactorily in the majority of published trials.42–45 Further experiences from the use of different targeted agents such as alpelisib or ipatasertib are anticipated.46,47
Among newer, emerging biomarkers, microRNAs (miRNAs) deserve mention. These small, non-coding RNAs play a role in gene expression and consequently influence proliferation, invasion, and cell differentiation.48
While their role in clinical practice has yet to be defined, several miRNAs have been identified as involved in gastric cancer.49,50 Circulating miRNAs have been proposed as a possible tool for the non-invasive diagnosis of gastric cancer and the detection of relapse.51–53 Long, non-coding RNAs, which, like miRNAs, represent non-translated RNA sequences, appear to be similarly involved in gastric cancer tumorigenesis and progression and may possess prognostic value.54–56 However, further studies are needed to determine their possible clinical use.
CONCLUSIONS
The treatment of GEA is no longer defined by a “one size fits all” approach, and the definition of molecular subgroups amenable to individualized treatment strategies is likely the way forward. However, the lack of standardization and the multiplication of proposed classifications and biomarkers represents a significant obstacle to the definition of a new paradigm. Furthermore, it is necessary to continue to study the heterogeneity of GEA: their genetic variations should drive investigations deeper into the molecular characteristics present in tumor tissue, and the use of circulating biomarkers to monitor disease evolution should be explored. While small-scale experiences are undoubtedly of value and essential for hypothesis generation, appropriately powered clinical trials are now urgently needed to translate these hypotheses in evidence-based practice.
TAKE-HOME MESSAGES
-
Multiple attempts are being made at classifying gastro-esophageal adenocarcinomas (GEAs) into molecular subtypes with prognostic or predictive value. These classifications often rely on the detection of multiple genetic and epigenetic alterations rather than a single, specific biomarker.
-
HER2 positive and MSI-H GEA benefit from biomarker-driven therapy that is part of clinical practice; angiogenesis-targeting strategies without biomarker selection are also part of standard management.
-
Interesting experiences in the tumor-agnostic setting may slowly be entering clinical practice (e.g., NTRK-directed therapies).
-
Many other biomarkers are still investigational as therapeutic targets; some of them have a proven prognostic role in the clinical setting.
-
Appropriately powered prospective clinical trials are now needed to provide evidence and to synthesize the growing number of small-scale experiences into a treatment paradigm that will benefit from the unraveling and classification of disease heterogeneity.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
All authors contributed to and approved the final manuscript.