
_ic_50_values_of_reversible_egfr-tkis_and_afatinib__b)_progression-free_survival_(pfs)_in.tiff)


INTRODUCTION
Over the past decade numerous large-scale sequencing approaches have identified increasingly more oncogenic driver mutations in non-small cell lung cancer (NSCLC) (Figure 1A),1 which have facilitated the rational development of targeted therapies and led to marked survival benefits compared to classical treatment approaches with chemotherapy (Figure 1B).2 Mutations in the epidermal growth factor receptor (EGFR) gene are amongst the most frequently observed,1,3,4 but significant differences in mutation frequency exist depending on histology, gender, smoking status and ancestry. The highest rate of EGFR mutations (≥50%) has been found in Asian female non-smokers with adenocarcinoma histology and a bronchioalveolar subtype.5–7
_distribution_of_oncogenic_driver_mutations_in_nsclc__b)_os_targeted_versus_non-targeted.png)
The EGFR family, also referred to as HER or ErbB receptors, comprises four receptor tyrosine kinases: EGFR (HER1), ERBB2 (HER2), ERBB3 (HER3) and ERBB4 (HER4). Ligand binding to the extracellular region of EGFR leads to autophosphorylation, activation and EGFR dimerization, which in turn initiates intracellular signaling cascades.5 Signal transduction subsequently leads to cell proliferation and survival via activation of RAS/RAF/MEK and PI3K/AKT pathways, among others.5 EGFR became a prime therapeutic target in NSCLC in 2004, following the discovery that somatic mutations in the EGFR gene enhance tyrosine kinase activity in lung cancer patients.5 These somatic EGFR mutations activate the kinase receptor in the absence of ligand binding, and can trigger oncogenesis by inducing a constitutively active state that leads to sustained downstream signaling.9 Subsequently, targeted therapy with tyrosine kinase inhibitors (TKIs) has emerged as the mainstay treatment for advanced NSCLC patients with activating mutations (Figure 1C).10
Three generations of EGFR-TKIs are currently approved but they are not all equal in terms of EGFR binding, metabolism, antitumor activity (Table 1A) and safety.11 First-generation EGFR-TKIs (e.g., gefitinib and erlotinib) reversibly bind to EGFR and inhibit the binding of adenosine triphosphate (ATP) to the tyrosine kinase domain (Figure 2A-B). Although gefitinib and erlotinib have shown efficacy in first-, second-, and third-line treatment of NSCLC, the benefit seen is usually transient because NSCLC with EGFR-activating mutations treated with first-generation EGFR-TKIs inevitably develop resistance.12 Up to half of patients treated with first-generation EGFR-TKIs develop acquired resistance through a T790M EGFR substitution mutation.13
_ic_50_values_of_reversible_egfr-tkis_and_afatinib__b)_progression-free_survival_(pfs)_in.tiff)
_egfr-tki-generations__and_b)_schematic_representation_of_egfr_signaling_and_egfr-tki_in.png)
Second-generation EGFR-TKIs (e.g., afatinib and dacomitinib) form irreversible, covalent attachments to the EGFR kinase domain and have demonstrated improvements in progression-free survival (PFS) relative to those treated with first-generation EGFR-TKIs.17,18 Moreover, the third-generation EGFR-TKI, osimertinib, is highly active in NSCLC patients with the T790M EGFR mutation who had disease progression with first- and second-generation EGFR-TKIs.19
Although the treatment landscape of advanced NSCLC has dramatically changed with the introduction of EGFR-TKIs, identifying methods to best select patients who are more likely to benefit from EGFR inhibition as well as selecting the optimal sequence of EGFR-TKIs, especially how best to use the third-generation EGFR-TKI, osimertinib, are emerging issues that still need to be addressed.
This critical review considers the evolving landscape of EGFR-TKI therapy and discusses the optimal sequence schedule to improve survival outcomes for EGFR-mutant positive (EGFR M+) advanced NSCLC patients.
FIRST-LINE EGFR-TKI THERAPY IN NSCLC
For almost a decade now, first- and second-generation EGFR-TKIs have been widely established in place of traditional standard platinum-based chemotherapy as the first-line treatment of choice for patients with newly diagnosed EGFR M+ advanced NSCLC.20
Several randomized trials have demonstrated that first-line EGFR-TKI therapy can improve objective response rates (ORR) and promote longer PFS compared to standard chemotherapy (Table 1B).18,21–28 Median PFS in phase III trials comparing gefitinib with platinum-based chemotherapy as first-line treatment of patients with advanced NSCLC is about 9 months versus 6 months, respectively.22,23 However, no overall survival (OS) benefit was identified in these first-generation EGFR-TKI trial populations, which may be partly due to high crossover rates following disease progression (Table 1C).10,18,21–28
EGFR MUTATIONS AND OUTCOMES WITH EGFR-TKIs
First-generation EGFR-TKIs were serendipitously found to be most effective in advanced NSCLC patients with tumors harboring recurrent activating mutations (mainly somatic)29,30 occurring in the exons encoding the kinase domain of EGFR.31–33 Approximately 93% of EGFR activating mutations occur in exons 19 to 21. Of these, deletions in exon 19 (Del19) and a point mutation in exon 21 leading to substitution of leucine for arginine at position 858 (L858R) are particularly common (44.8% and 39.8%, respectively).34 Indeed, the presence of an EGFR mutation in NSCLC patients is the strongest and most reliable predictor of improved PFS and ORR with EGFR-TKIs compared with chemotherapy in the first-line setting. The 2009 landmark IPASS (Iressa Pan-Asia Study) study reported significantly longer PFS with gefitinib versus standard chemotherapy in a subgroup of advanced lung tumor patients with EGFR mutations (hazard ratio, [HR] for progression to death: 0.48 [95% CI: 0.36–0.64]; p<0.001) but not in patients with EGFR wild-type tumors (HR for progression to death: 2.85 [95% CI: 2.05–3.98]; p<0.001).35
Much later in 2015, a pooled analysis of the LUX-Lung 3 (LL3) and LUX-Lung 6 (LL6) trial populations showed superiority of afatinib to platinum-based chemotherapy in terms of median OS in a subset of patients with EGFR Del19 mutations but not in patients with EGFR L858R mutations: 27.3 versus 24.3 months, respectively (HR: 0.81 [95% CI: 0.66–0.99]; p=0.037).36 This data revealed for the first time that EGFR-TKIs in the first-line setting could prolong OS in patients with EGFR L858R mutations: 27.3 versus 24.3 months, respectively (HR: 0.81; [95% CI: 0.66 to 0.99]; p=0.037).36 This data revealed for the first time that EGFR-TKIs in the first-line setting could prolong OS in patients harboring Del19 EGFR mutations, and that Del19 and L858R EGFR mutations constitute two distinct patient subgroups.36
In a small proportion of patients, primary resistance mutations can already be detected at diagnosis (Figure 3). The most prevalent are EGFR exon 20 insertions (4%) and EGFR T790M mutations (5%).1,3,4,8,37 While the latter are sensitive to third-generation EGFR-TKIs, EGFR exon 20 insertions can be targeted with the specific inhibitor TAK-788.38 Initial results from a phase I/II trial (NCT02716116) showed a disease control rate (DCR) of 89% with 54% of the patients achieving a partial response (confirmed + unconfirmed) and 35% stable disease. Overall, 23 of 24 evaluable patients (93%) showed a reduction in the tumor volume; side effects were consistent with other EGFR-TKIs. Therefore, a phase III trial (n=318, 1:1 randomization) comparing TAK-788 with platinum/pemetrexed combination chemotherapy was initiated with enrolment starting in late 2019 (NCT04129502).38 Furthermore, in March 2020 JNJ-61186372, a bi-specific antibody targeting EGFR and c-MET, received FDA breakthrough designation for the poor-prognosis subpopulation of EGFR M+ patients harboring EGFR exon 20 insertions. This status was granted after promising phase I results. JNJ-61186372 will be further evaluated in combination with lazertinib.39
_nsclc.png)
CO-MUTATIONS AND OUTCOMES WITH EGFR-TKIs
Although EGFR mutant NSCLC represents a prototypical oncogene-addicted cancer with a single driver, co-mutations are a frequent event.40 The most commonly mutated genes are TP53 (55–65%), RB1(10%), PI3KCA/PTEN (9–12%) and CTNNB1 (5–10%).1,41 TP53 alterations are associated with a poorer prognosis (lower ORR, shorter PFS and OS).41,42 Similar findings have been made for PTEN, MDM2 and RB1 mutations, especially for patients with EGFR mutations and RB1 loss; PFS with EGFR tyrosine kinase inhibition was extremely short (median PFS of 1.9 months).41 Alterations in TP53, RB1, and the combination of both in particular, are associated with increased genetic instability eventually leading to small cell transformation.41,42
SECOND-GENERATION VERSUS FIRST-GENERATION EGFR-TKIs
Recent head-to-head trials have demonstrated that second-generation EGFR-TKIs are preferable to first-generation EGFR-TKIs as first-line treatment for patients with EGFR M+ NSCLC since they may prolong PFS and OS. First-line efficacy of erlotinib or gefitinib has been compared with second-generation EGFR-TKIs in randomized clinical trials. In the LUX-Lung 721 and ARCHER-1050 trials,18 gefitinib was compared with second-generation agents afatinib and dacomitinib, respectively. The LUX-Lung 7 was a large (N=319), multicenter and multi-ethnic phase IIb trial that showed a trend toward improved OS with afatinib versus gefitinib in treatment-naïve patients harboring Del19 or L858R mutations.21 Overall, analysis of the LUX-Lung 7 data together with the LUX-Lung 3 and LUX-Lung 6 data show that afatinib results in approximately 10% of patients achieving long-term clinical benefit (long-term response ≥3 year PFS and OS), which is greater than that observed with first-generation EGFR-TKIs.17 Stratification factors in these trials included: Del19 versus L858R EGFR and versus other ‘uncommon’ mutations in LUX-Lung 3 and LUX-Lung 6; race (Asian vs non-Asian) in LUX-Lung 3 only, and brain metastases (presence vs absence) in LUX-Lung 7 only.17 The frequency of Del19 mutation-positive NSCLC was slightly higher among long-term responders in the LUX-Lung 3, 6, 7 analysis, which supports its known role as a biomarker for improved outcomes with EGFR-TKIs versus other mutation types.17 These studies also demonstrate that afatinib can provide clinical benefit in advanced NSCLC patients with brain metastases or uncommon mutations.21,26,27 Although the LUX-Lung 7 trial showed that afatinib compared to gefitinib significantly improves PFS and the time-to-treatment failure, more serious drug-related adverse events were reported in the afatinib group than in the gefitinib group.43 Indeed, second-generation EGFR-TKIs typically have more frequent side effects than other EGFR-TKIs because of their broad inhibitory profile.11 However, post-hoc analyses from LUX-Lung 3 and LUX-Lung 6 suggest that tolerability-guided dose adjustment of afatinib is an effective measure to reduce treatment-related adverse events, as well as reduce interpatient variability of afatinib exposure, without affecting treatment efficacy.44 A more recent study by Yokoyama et al. (2019) shows that a low starting dose of afatinib therapy (20 mg daily dose) with dose modification according to severity of adverse events is a better strategy in treatment-naïve patients who have NSCLC associated with common activating EGFR mutations, both in terms of effectiveness and tolerability, than starting with a standard afatinib dose (40 mg daily).45 In this phase II trial, the PFS was 15.2 months, which was similar to previous reports with the standard 40 mg dose of afatinib, and toxicities were generally mild. Importantly, the low starting dose of afatinib was also effective in high-risk patients with common EGFR M+ NSCLC who had asymptomatic brain metastasis.45
The ARCHER 1050 trial is the first randomized, phase III study that directly compared a second-generation versus a first-generation EGFR-TKI to demonstrate an OS benefit.46 Although, in contrast to the LUX-Lung 3,26 6,27 and 721 trials, the ARCHER 1050 trial identified a significant improvement in OS with dacomitinib versus gefitinib (median OS: 34.1 vs 26.8 months, respectively; HR: 0.76 [95% CI: 0.582–0.993]; p=0.044), it did not include patients with uncommon mutations or brain metastases.46
ACQUIRED RESISTANCE MECHANISM TO FIRST- OR SECOND-GENERATION EGFR-TKIs
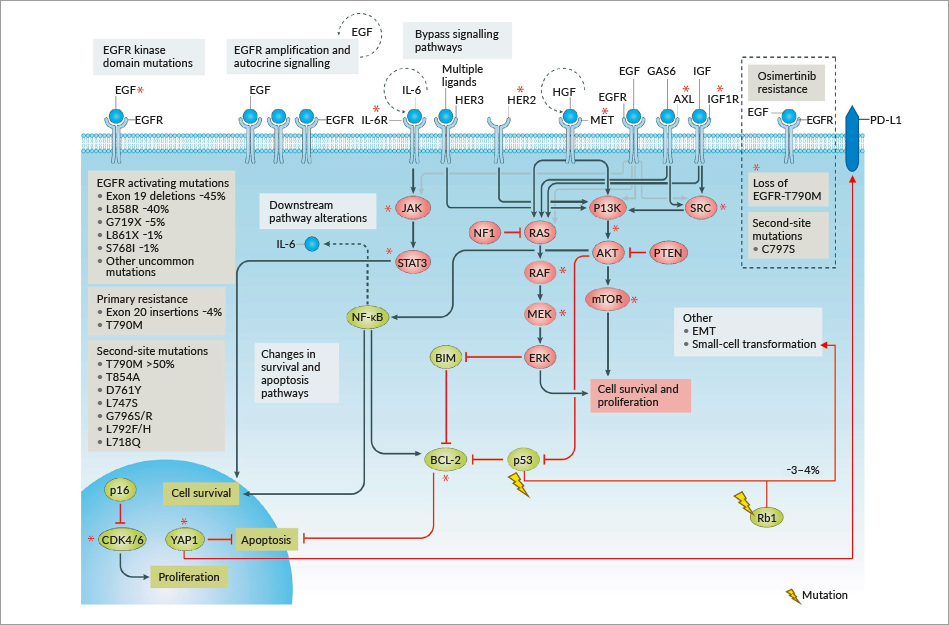
The majority of NSCLC patients who harbor EGFR activating mutations show an initial pronounced response to EGFR-TKI treatment, but ultimately acquire resistance to these drugs after approximately 9 to 14 months of therapy.47 The threonine 790 to methionine (T790M) secondary mutation in exon 20 of EGFR is the most common mechanism conferring acquired resistance; it is detected in approximately 50% of patients treated with first-generation EGFR-TKIs.13 Several other EGFR-dependent and EGFR-independent mechanisms of acquired resistance have been described, including secondary site EGFR mutations (T290M, C797S), mesenchymal-epithelial transition factor (MET) amplification, human receptor tyrosine kinase epidermal growth factor receptor-2 (HER2) amplification, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) mutations or amplification, phosphatase and tensin homolog (PTEN) loss, mitogen-activated protein kinase (MAPK) pathway activation, BRAF mutation, insulin-like growth factor receptor-1 (IGF-1R), fibroblast growth factor receptor (FGFR) activation, and small cell lung cancer (SCLC) transformation, among others.48–50 It has been proposed that the common T790M resistance mutation reduces the potency of ATP-competitive kinase inhibitors and that irreversible inhibitors could overcome this resistance via covalent binding, rather than an alternative binding mode.51 Amplification of MET occurs in approximately 5% to 20% of patients whereas HER2 amplification is estimated to occur in up to 8% of patients.52 Lineage plasticity, specifically small cell histologic transformation, occurs in 5%–14% of patients with resistance to earlier generation EGFR inhibitors.53,54
Second-generation irreversible EGFR-TKIs were initially designed and developed to delay or overcome T790M-mediated resistance, however evidence from the literature indicates a similar frequency of T790M-mediated resistance in patients receiving first-line treatment with the second-generation EGFR-TKI afatinib.55,56 Despite a delay in the expression of T790M when used in the first-line setting, the T790M mutation is still detected in 36.4% or 47.6% of patients treated with afatinib.55,56 Although preclinical data showed promising results of afatinib against EGFR T790M,57 the clinical trial data was disappointing, showing no OS benefit in patients after failure of platinum doublet chemotherapy and first-generation EGFR-TKIs.58 Notably, T790M is rarely identified in tumors before exposure to EGFR-TKIs concurrently with other more common activating mutations.48
Presently, the clinical efficacy of dacomitinib and osimertinib against uncommon mutations is being evaluated in ongoing phase II trials.59 In addition, an exploratory phase II trial (ZENITH20) with poziotinib, a new pan-HER TKI, in NSCLC patients harboring exon 20 insertions failed to reach its primary endpoint in a cohort of 115 patients.60
Patients with wild-type EGFR may present concurrent oncogenic mutations in KRAS proto-oncogene GTPase (KRAS), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α (PIK3CA), and tumor protein p53, resulting in differential clinical features, treatment outcomes and survival prognoses.61 Zhao et al (2019) showed that patients with the wild-type genes experienced longer PFS times than those with KRAS, TP53, KRAS/TP53 or PIK3CA/TP53 mutations.61 Moreover, the authors suggest that patients with NSCLC should receive routine KRAS, PIK3CA and TP53 gene sequencing to determine mutations for the analysis of clinical characteristics and prognosis.61 As mentioned above, these mutations may be detected as part of the initial mutational spectrum where they mediate primary resistance, but they may also emerge during the course of EGFR-TKI treatment (i.e., secondary resistance) (Figure 3).1
EXTENDING SURVIVAL WITH THIRD-GENERATION EGFR-TKIs IN TREATMENT-NAÏVE NSCLC PATIENTS
Osimertinib is an irreversible, third-generation EGFR-TKI that was developed based on its ability to inhibit the T790M resistance mutation and its selectivity for the mutant receptor. Since it also inhibits the common EGFR-activating mutations, namely Del19 and L858R, its role as first-line therapy represents a new milestone for patients with EGFR mutations.10,19
In the recent phase III FLAURA study, median OS was positive for osimertinib compared to older generation EGFR-TKIs in patients with Del19 or L858R EGFR mutated advanced NSCLC (38.6 months vs 31.8 months; HR for death, 0.80 [95.05% CI: 0.64 to 1.00]; p=0.046).62 In the first-line setting when compared with gefitinib or erlotinib, osimertinib met its primary endpoint showing significant improvement in PFS (10.2 vs 18.9 months, respectively; HR: 0.46 [95% CI: 0.37–0.57]; p<0.001)10 and OS (31.8 vs 38.6 months; HR: 0.8 [95% CI: 0.641–0.997]; p=0.0462).62 The superior PFS results with osimertinib were consistent across common types of EGFR mutations, namely Del19 and L858R, and against patients with or without baseline brain metastases. Osimertinib is also highly active against confirmed metastatic or recurrent NSCLC with uncommon activating EGFR mutations, e.g., mutations other than Del19, L858R, T790M and exon 20 insertions; notably, 63% of patients in this study received first-line osimeritinib.63 Taken together these data suggest that osimertinib is more effective and better tolerated than first-generation EGFR-TKIs, and thus osimertinib monotherapy has emerged as the new standard-of-care for first-line treatment of EGFR-mutated NSCLC patients.10,62 Notably, however, FLAURA compared osimertinib with either gefitinib or erlotinib but not with afatinib.64
STRATEGIES TO FURTHER IMPROVE SURVIVAL OUTCOMES FOR NSCLC PATIENTS IN THE FIRST-LINE SETTING
In addition to monotherapy, EGFR-TKIs in combination with chemotherapy65 or immunotherapy66,67 have shown improvement in PFS in patients with advanced NSCLC harboring EGFR mutations.
One strategy that attempts to decrease the emergence of resistance involves combining cytotoxic chemotherapy with an EGFR-TKI.68,69 A recent study by Noronha et al. (2019) demonstrated that first-line gefitinib combined with pemetrexed and carboplatin chemotherapy significantly improved PFS compared with gefitinib alone (16 months vs 8 months [95% CI: 7.0–9.0 months], respectively; HR for disease progression or death: 0.51 [95% CI: 0.39–0.66]; p<0.001) and OS (not reached vs 17 months [95% CI: 13.5–20.5 months]; HR for death: 0.45 [95% CI: 0.31–0.65]; p<0.001), but increased toxicity in patients with NSCLC.68 However, results with the first-generation EGFR-TKI and pemetrexed as first-line treatment for NSCLC does not exceed osimertinib monotherapy in terms of PFS in this setting.10,68,70 Moreover, grade ≥3 toxicities occurred in 34% and 51% of patients in the osimertinib trial versus the gefitinib plus chemotherapy trial, respectively.10,70
Results from the randomized phase III RELAY trial showed that in the first-line setting erlotinib in combination with ramucirumab reduces the risk of disease progression or death by over 40% versus erlotinib alone in EGFR-positive NSCLC.71 At a median follow-up of 20.7 months, the median PFS was 19.4 months (95% CI: 15.4–21.6) with the ramucirumab combination compared with 12.4 months (95% CI: 11.0–13.5) with erlotinib alone (HR: 0.591 [95% CI: 0.461–0.760]; p<0.0001).71
Bevacizumab is a promising antibody for blocking vascular endothelial growth factor receptor 2 (VEGFR2). It is being evaluated in combination with EGFR-TKI as a first-line treatment for NSCLC in clinical trials.72 Compared with erlotinib alone, bevacizumab plus erlotinib combination therapy improved PFS in patients with EGFR-positive NSCLC. This study is ongoing and further follow-up is required to assess OS and overall efficacy of this combination in the first-line setting.66
TREATING NSCLC PATIENTS WITH UNCOMMON EGFR MUTATIONS
Uncommon mutations may occur singularly or co-exist with a common or another uncommon EGFR mutation.73 Up to 25% of all EGFR mutation-positive tumors may have uncommon mutations together with a common EGFR mutation within the same tumor.73–75 Moreover, individuals with EGFR co-mutations exhibit shorter PFS and lower response rates than those with single EGFR mutations.76 The substitution mutations of G719X in exon 18, L861Q in exon 21, S768I in exon 20, and exon 20 insertions are most prevalent among uncommon mutations described to date.77 Patients with these uncommon substitution mutations benefit from first-generation EGFR-TKIs such as erlotinib and gefitinib,78–80 however, based on extensive and robust data, afatinib is the preferred agent for the first-line treatment of NSCLC harboring these mutations.74
Afatinib has demonstrated clinical activity against G719X, L861Q and S768I in a post-hoc analysis of the LUX-Lung trials81 as well as in the real-world.74 Of 75/600 (12%) NSCLC patients with uncommon EGFR mutations in a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6 clinical trials, the ORR was 78% with a PFS of 13.8 months for G719X (n=14), 56% with a PFS of 8.2 months for L861Q (n=9), and 100% with a PFS of 14.7 months for S768I (n=8).36 In a recent large, real-world dataset identifying patients with 98 different uncommon mutations, the ORR with afatinib was 60% in EGFR-TKI naïve patients (G719X: 63.4%; L861Q: 59.6%; and S768I: 62.5%) and 25% in EGFR-TKI pretreated patients.71 Seven (6.8%) EGFR-TKI naïve patients in the real-world dataset continued with afatinib treatment for more than three years; this is only marginally lower than reported in clinical trials (10–12%).17 Furthermore, whilst exon 20 insertions are traditionally not considered responsive to EGFR-TKIs based on retrospective analyses,80,82,83 analysis of the aforementioned real-world dataset demonstrated that some exon 20 insertions are clinically sensitive to afatinib; analysis of 70 NSCLC patients with exon 20 insertions treated with afatinib showed an ORR of 24.3% with a median duration of response (DoR) of 11.9 months.74
In a preclinical study by Kohsaka et al. (2019), it was shown that afatinib, possibly due to its alternative mechanism of action, reduced the viability of cells expressing numerous compound mutations, including those involving Del19, G719X and/or S768I mutations; only compound mutations involving T790M were considered to be resistant to afatinib.84
SECOND-LINE TREATMENT OPTIONS FOR NSCLC PATIENTS AND THE NEED FOR SENSITIVE T790M DETECTION ASSAYS
Osimertinib monotherapy has demonstrated superior activity versus platinum-pemetrexed chemotherapy as second-line treatment for EGFR T790M M+ NSCLC and is the current standard treatment in this setting.19,85 In addition, afatinib and osimertinib have been shown to inhibit EGFR phosphorylation in cells harboring uncommon secondary resistance mutations.86–89 For patients without EGFR T790M mutations, platinum-based chemotherapy is the currently recommended second-line treatment.90–92 Unlike the emergence of EGFR T790M in response to earlier generation EGFR-TKIs, there is no predominant mechanism of resistance to target on progression to osimertinib.52 Hence, there is no established targeted treatment following the failure of osimertinib, so it is widely debated whether osimertinib should be reserved for second-line use following progression with a second-generation EGFR-TKI.93
In the confirmatory phase III (AURA 3) trial, osimertinib was superior to platinum-pemetrexed in patients with T790M-positive advanced NSCLC, who had disease progression after first- or second-generation EGFR-TKI therapy.19 The median PFS was 10.1 months in the osimertinib arm and 4.4 months in the platinum-pemetrexed arm (HR: 0.30 [95% CI: 0.23–0.41]; p<0.001). The ORR was significantly better with osimertinib (71% [95% CI: 65–76]) than with platinum-pemetrexed (31% [95% CI: 24–40]) (odds ratio: 5.39 [95% CI: 3.47–8.48]; p<0.001). The median duration of response was 9.7 months (95% CI: 8.3–11.6) with osimertinib and 4.1 months (95% CI: 3.0—5.6) with platinum-pemetrexed.19 At data cut-off, the median OS was 26.8 months (95% CI: 23.5–31.5) versus 22.5 months (95% CI: 20.2–28.8) respectively (HR: 0.87 [95% CI: 0.67–1.12]; p=0.277); the survival rate at 24 months was 55% versus 43% and at 36 months was 37% versus 30%, respectively.94
In a real-world clinical practice setting, EGFR-TKI-naïve patients who received first-line afatinib and went on to develop T790M-positive acquired resistance subsequently received second-line osimertinib.93 At the initial database lock the median time to treatment failure (TTF), the primary outcome, was 27.6 months (90% CI: 25.9–31.3 months).95 Results were also promising in patients with an EGFR Del19 activating mutation (30.3 months) and Asian patients (46.7 months).95 An interim analysis has demonstrated a median OS of 41.3 months (90% CI: 36.8–46.3) overall and 45.7 months (90% CI: 45.3–51.5) in patients with Del19-positive tumors (n=149).96 The 2-year survival was 80% and 82%, respectively, for patients overall and those with Del19 tumors.96
A key challenge of the second-line use of osimertinib is to ensure that all patients who develop T790M-driven resistance are identified; this requires re-biopsy, and the widespread availability of sensitive T790M detection assays. EGFR mutations can be analyzed in tumor biopsies and in cell-free plasma DNA from blood samples (liquid biopsy).97,98 Selection of EGFR-mutated NSCLC patients for osimertinib treatment using liquid biopsy supported by tumor tissue analyses in plasma-negative cases may now be the preferred strategy.97,98 Highly sensitive genotyping assays have also been developed to use with liquid biopsy samples, e.g., the droplet digital polymerase chain reaction (ddPCR) can reliably detect mutations with high sensitivity and specificity.97,98 In a recent comparative study, ddPCR was more sensitive than Cobas® EGFR Mutation Test v2 for detecting EGFR T790M mutations in cell-free plasma DNA.98 Notably, in patients who progressed under treatment with an EGFR-TKI, the T790M positivity rate was 66% using ddPCR, but only 24% using Cobas.98 Overall, in this real-world setting, 60% of all T790M-positive patients, as assessed by ddPCR and Cobas®, were subsequently treated with osimertinib.98
COMBINATION STRATEGIES
Studies with other exploratory second-line treatments are ongoing. For example, activity of EGFR-TKI and anti-EGFR-antibody, chemotherapy99,100 and immunotherapy101 combinations are being explored in the second-line NSCLC setting. Exploratory combinations with immunotherapies such as programmed death ligand-1 (PD-L1) inhibitors have been generally disappointing, i.e., they have failed to demonstrate a clinical benefit, plus they have been limited by the toxicity rates leading in many instances to treatment discontinuation;102 only the IMPOWER150 study, evaluating the combination of carboplatin, paclitaxel and bevacizumab plus or minus the PD-L1 inhibitor, atezolizumab, has shown encouraging results to date.103,104
EGFR inhibition combined with afatinib and the monoclonal anti‐EGFR antibody, cetuximab, showed a beneficial response in resistant tumors harboring the EGFR T790M mutations in a phase Ib clinical trial.105 This trial included advanced EGFR M+ NSCLC patients with acquired resistance to erlotinib or gefitinib who were administered afatinib plus cetuximab.105 The ORR in the T790M-positive and negative subgroups were comparable (32% vs 25%; p=0.341), while the median duration of response and median PFS were 5.7 and 4.7 months, respectively.105 Moreover, afatinib-cetuximab demonstrated a manageable safety profile in this setting.105 Interestingly, triplet therapy, including afatinib, cetuximab, and bevacizumab induced pathological complete remission (CR) repeatedly in lung cancer cells harboring EGFR T790M mutations in vivo.106
Combination strategies with other TKIs such as BRAF, MEK and MET TKIs are also being evaluated. Crizotinib, for example, is a multitargeted MET/ALK/ROS1 inhibitor that is currently used as a MET-inhibitor; notably ALK mutations are found at low frequency in NSCLC (approximately 5%).107 Several case reports have already described the successful use of osimertinib combined with dabrafenib plus trametinib following an acquired BRAF V600E mutation,108,109 or osimertinib combined with crizotinib for EGFR M+ NSCLC with a treatment-emergent MET amplification.110 The TATTON multi-arm phase Ib study is the first trial to report the safety and efficacy of the combination osimertinib plus savolitinib in pretreated EGFR M+, MET amplified NSCLC.111 Response rates up to 67% and a median PFS of up to 11.0 months (depending on mutational profile and preceding therapy) together with an acceptable safety profile were observed.111
Despite the lack of an overall benefit, the randomized phase II trial evaluating the c-MET monoclonal antibody emibetuzumab is a landmark trial.112 The trial suggests that the negative prognostic impact of a biomarker (c-MET overexpression) can be completely reversed by a rational, biomarker-driven combination, which turns into a predictive biomarker at the same time.112 An exploratory post-hoc analysis showed an improvement of 15.3 months in median PFS (20.7 with emibetuzumab plus erlotinib versus 5.4 months with erlotinib (HR: 0.39 [90% CI: 0.17–0.91]) for the 24 patients with the highest MET expression (MET expression level of 3+ in ≥90% of tumor cells).112
THIRD- AND SUBSEQUENT-LINE TREATMENT OPTIONS
Results from a retrospective analysis of subsequent therapy outcomes in patients with common EGFR mutations in LUX-Lung 3, 6 and 7 trials supported treatment sequencing with first-line afatinib followed by subsequent therapies, including first-generation EGFR-TKIs and osimertinib (n=37; mostly in the ≥ third-line setting; 10 patients received second-line osimertinib). The median duration of osimertinib therapy in any treatment line was 20.2 months (95% CI: 12.8–31.5). Median PFS on afatinib in 37 patients receiving subsequent osimertinib was 21.9 months; median OS was not reached after a median follow-up of 4.7 years.113 Notably, only a few patients received osimertinib following afatinib because the availability of osimertinib at the time these LUX-Lung studies were undertaken was restricted, plus testing for T790M was not required or documented.113
EFFICACY OF EGFR-TKIs IN BRAIN METASTASES AND LEPTOMENINGEAL DISEASE
About 20–40% of patients with NSCLC develop brain metastases (BM) during the course of their disease and historically their prognosis is generally poor.114,115 The median survival of patients with untreated brain metastases is reported to be 1–3 months.116–118 Moreover, approximately one-third of NSCLC patients develop central nervous system (CNS) metastases after acquiring EGFR-TKI resistance.119,120 Despite preclinical data suggesting poor blood-brain-barrier (BBB) penetrance,121 EGFR‐TKIs have demonstrated systemic efficacy and CNS activity in patients with BM.122
Studies have shown that EGFR-TKIs, namely afatinib and osimertinib, are able to cross the BBB.122,123 For example, Tamiya et al. (2018) investigated the efficacy of afatinib in a prospective study of 11 patients with EGFR-mutated NSCLC with leptomeningeal carcinomatosis.122 The median cerebrospinal fluid concentration of afatinib in this study was 3.16±1.95 nM.122 This equates to a median concentration of 2.9 nM, which is clearly above the IC50 value for the EGFR (0.5 nM).122,124
The Cambridge Brain Mets Trial 1 (CamBMT1) was the first proof-of-principle trial examining the concentration of afatinib in operable BM of patients treated with afatinib for 11 days plus irradiation (2 or 4 Gy) on day 10 prior to resection.125 The phase Ib results of the 10 patients investigated in the phase Ib study showed that in the resected BM, the concentration of afatinib was a median of 405 ng/g, i.e., more than 15 times higher than in the plasma (median 22.7 ng/mL).125 The subsequent phase II study is still ongoing, with data from a total of 60 patients being investigated.125 CNS efficacy has also been demonstrated in several subanalyses of clinical studies.126 For example, in a pre-specified subanalysis of the LUX-LUNG 3 and 6 studies, it was shown that the extent of PFS benefit with afatinib versus first-line chemotherapy in patients with EGFR-mutated NSCLC was comparable between patients with and without brain metastases.126 In a combined analysis, afatinib significantly prolonged PFS compared to chemotherapy in patients with brain metastases (8.2 vs 5.4 months, respectively; HR: 0.50; p=0.0297).126
Recent additional analyses of the LUX-LUNG 3 and 6 studies have shown that patients with CNS involvement on afatinib had a lower risk of progression in the CNS than patients with progression elsewhere.127 Likewise, the risk of CNS progression de novo under afatinib was low; observed in only 6% of patients without CNS involvement at baseline.127 Data are also available supporting the activity of afatinib in the CNS for pretreated patients with EGFR M+ NSCLC, and with at least two previous lines of EGFR-TKI plus platinum-based chemotherapy.14 Hoffknecht et al. (2015) showed that the median time to treatment failure of 3.6 months was comparable between patients with and without CNS metastases. In addition, cerebral disease control was achieved in 66% of patients with CNS involvement.14 This data is also supported by a number of cases from daily clinical practice.128 Hochmair et al. (2016), for example, report on five patients with symptomatic brain metastases in whom a complete remission could be achieved under afatinib, which according to magnetic resonance imaging (MRI) lasted for at least five months and was accompanied by a correspondingly large clinical benefit.128 Most recently, Gottfried et al. (2019) presented data demonstrating the efficacy of afatinib in patients, both Asian and non-Asian, with BM.129 The median time to symptomatic progression (TTSP) was 13.7 months in this combined real-world data analysis, which shows consistency with data from the phase III LUX-Lung 3 and LUX-Lung 6 studies.129
The AURA studies have demonstrated CNS activity of osimertinib in pre-specified subgroup analyses of patients with EGFR T790M-positive NSCLC who had progressed while on previous EGFR-TKI treatment.130,131 Moreover, the consistent CNS efficacy observed across analyses in the FLAURA study, provides strong evidence for the superior CNS efficacy of osimertinib.10
Leptomeningeal disease is another CNS metastatic manifestation involving the leptomeninges and cerebrospinal fluid (CSF). Among EGFR M+ NSCLC patients, leptomeningeal metastases (LM) occur in approximately 9% of cases, which is double the 3.8% incidence in the overall NSCLC population.132 The prognosis for EGFR M+ NSCLC with LM is very dismal with a median OS ranging between 3 and 10 months from the time of diagnosis.133–135 This can be partially explained by the diffuse nature of the disease, making it less accessible for local interventions like surgery or radiotherapy, which are typically applied in emergency situations or only when patients are very symptomatic.136
In the BLOOM phase I trial (n=41) osimertinib was evaluated in patients with LM from EGFR-mutated advanced NSCLC whose disease had progressed on previous EGFR-TKI therapy. Yang et al. (2020) reported an ORR of 62% with a median DoR of 15.2 months.137 Osimertinib further led to an improvement of neurological symptoms and CSF clearance in 57% and 28% of the patients, respectively.137 Notably, the median OS of 11.0 months slightly surpassed historically reported results.137
DISCUSSION
The overall survival benefit observed in the FLAURA trial is an important milestone in EGFR M+ NSCLC. The improved PFS and the OS benefit of >6 months10,138 has led to Level I/Grade A recommendations in international guidelines.90,91,139 But does this make osimertinib the standard of care for every patient or just one of many different options? From the subgroup analysis of the FLAURA trial there seems to be no clinical or molecular determinant showing any detrimental outcomes of osimertinib compared to first-generation TKIs.138 On the other hand, the observed OS benefits seem to be mainly driven by, or at least more pronounced in, non-Asian patients with EGFR Del19 mutations and WHO performance status 1 plus detectable EGFR mutations in circulating tumor DNA (ctDNA).138 The detection of EGFR genetic alterations in ctDNA seems to be a poor-risk factor at any stage of the disease (shorter PFS when initially detected and shorter OS at relapse).140 Since the OS is not only influenced by the efficacy of the first-line treatment but also by subsequent treatments, it is worth looking at the reported data on factors changing the effectiveness of sequential therapy lines.
There seem to be no obvious differences in the FLAURA trial in terms of patients who received no subsequent second-line treatment due to death (22% in both arms) or patients alive not receiving any treatment (9% osimertinib arm [n=279] and 8% comparator arm [n=277]).138 The higher rate of patients still on study treatment in the osimertinib arm (22% vs 5%) also explains the lower rate of patients receiving a second-line treatment (48% in the osimertinib vs 65% in comparator arm).138 Second-line treatments mainly consisted of cytotoxic chemotherapy (68%) in the osimertinib arm, while in the comparator arm the majority of patients crossed over to osimertinib (47%) or received another EGFR-TKI (27%).138
Although no data are available to date on the second-line treatment outcomes, the numbers reflect the rate of mutations (approx. 50%) after first-generation TKI therapy, as reported in previous trials.13,47 Third-line therapy was consistent in both arms in terms of frequency (54% osimertinib arm vs 51% comparator arm) and composition.138 It is therefore highly unlikely that the estimated OS benefit is biased by lower subsequent treatment rates or the lack of accessibility to novel treatments.
No conclusions can be drawn for second-generation TKIs like afatinib36 or dacomitinib46, where improvements in progression-free and overall survival were observed as well. The results of the GioTag database96 and pooled post-hoc analysis of the LUX-LUNG 3, 6 and 7 trials113 suggest that the sequence afatinib followed by osimertinib provides excellent outcomes with a median OS of 41.3 months in the GioTag study and an OS that was not reached in LUX-LUNG 3, 6 and 7 after a median follow-up of 4.7 years (Figure 4a).
_overall_survival_of_sequential_therapy_for_afatinib_followed_by_osimertinib_in_the_real.png)
The crux of the matter is that these analyses also excluded patients with a poor prognosis, i.e., patients without second-line therapy or a EGFR T790M negative progression. Therefore in our opinion, the sequence afatinib followed by osimertinib can only be justified if we can better predict the type of progression and if we have OS data including afatinib patients receiving alternative second-line therapy (e.g., chemotherapy, immunotherapy, chemoimmunotherapy, or no therapy). The issue remains another important and debatable point,141 especially when the short PFS of 2.8 months with osimertinib as second-line treatment (AURA trial) is taken into account (Figure 4B).53 Notably, efficacy of second-line chemotherapy seems to be unaffected by the type of resistance (EGFR T790M negative vs EGFR T790M positive).142
Pictured is the sequencing strategy in which the second-generation EGFR-TKI, afatinib, is used in the first line, followed by the later-generation EGFR-TKI osimertinib after progression in the setting of acquired T790M mutation. Progression-free survival (PFS) for first-line afatinib is drawn from the LUX-LUNG 3, 6 and 7 trial data, which compared afatinib to conventional chemotherapy or gefitinib (LUX-LUNG 7) in the first-line setting. The median PFS for osimertinib after progression on afatinib in T790M-positive patients was derived from the GioTag trial. The median PFS for osimertinib and atezolizumab after progression on EGFR-TKIs in T790M-negative patients was derived from the AURA and IMPOWER150 trials, respectively. The lower picture is the sequencing strategy in which the third-generation TKI, osimertinib, was used as a first-line treatment based on the PFS demonstrated in the FLAURA trial followed by atezolizumab based on the PFS demonstrated in the IMPOWER150 trial. This figure illustrates that the superior efficacy of third-generation therapies in the front line may not lead to superior overall survival despite a clearly superior PFS when comparing front-line therapies in isolation.
It also remains an open question of how to best integrate immune checkpoint inhibitors into the treatment algorithm of EGFR M+ NSCLC. The IMPOWER150 phase III trial has shown an impressive OS benefit for the combination of atezolizumab, bevacizumab, carboplatin and paclitaxel in patients with at least one prior TKI treatment.104 However, the US Food and Drug Administration (FDA) label for this active regimen is restricted to patients who have exhausted other FDA-approved therapies.143 According to the European Medicines Agency (EMA) label, the regimen is indicated only after failure of appropriate targeted therapies.144 Since combination trials with osimertinib and durvalumab have been terminated due to severe side effects,145 it will be important to answer the question of when immune checkpoint inhibitors are optimally given, i.e., before or after a third-generation TKI. Although responses to PD-(L)1 monotherapy are generally low, outcomes seem to differ between EGFR Del19 and EGFR L858R NSCLC.146 The lowest response rates (7%) along with a significantly shorter PFS and OS were reported for EGFR Del19.146
Hastings et al. (2019) tried to explain these findings in relation to a lower mutational burden in EGFR Del19 NSCLC, which the authors subsequently observed in this subgroup of patients.146 In-vitro studies also indicate a direct immunosuppressive effect of dendritic cells mediated by exosomes of EGFR Del19 cells.147 Together, these results suggest that further stratification of EGFR M+ NSCLC might allow a better patient stratification and selection for immune checkpoint inhibitor treatment.
Whether the use of chemotherapy plus TKI combinations is justified remains another matter of debate. After trials in molecularly unselected patients have failed to demonstrate any benefit, two recent trials have rekindled the discussion.148 Although a study by Noronha et al. (2019) has shown a PFS and OS benefit, it has been criticized that the PFS (16 months) and OS (not reached after a median follow-up of 17 months) does not exceed the results of the FLAURA trial.149 The fact that only 15% received a third-generation EGFR-TKI was seen as another methodological issue.70 Therefore, the increased toxicities might not be justified when other modern treatment options are available.70 Despite the comparably low rate of third-generation EGFR-TKI treatment (22%), the PFS (20.9 months) and OS (50.9 months) in the NEJ009 phase-III trial is unprecedented.69 Whether the infrequent application of third-generation EGFR-TKIs in the above mention trials might be explained by a different mutational spectrum at progression is still an open question. It will be also important to answer if co-mutations or certain mutational profiles will be able to identify patients who are able to obtain an extraordinary benefit through the addition of chemotherapy. In this context the results of the FLAURA II trial (NCT04035486) combining osmertinib plus platinum/pemetrexed are highly anticipated.
Finally, treatment beyond progression along with the integration of local interventions like radiotherapy is another important question to be discussed, especially for patients with oligo-progressive disease. An observational study with 577 EGFR M+ patients suggested that treating patients beyond progression does not negatively impact survival.150 Smaller trials and retrospective analyses suggested that radiotherapy (RT) or multi-site stereotactic body radiotherapy (SBRT) is able to induce responses (50% ORR, >80% local DCR) and extend the PFS about 4–10 months, delaying the change of second-line treatment.151–153 The combination of radiotherapy and treatment beyond progression might even extend OS, as suggested in the retrospective analysis of 118 patients cohorts from two cohorts from the University of Texas MD Anderson Lung Cancer Moonshot GEMINI and Moffitt Cancer Center lung cancer databases.154 Furthermore, a randomized phase III trial (n=300, NCT03944772) started to recruit patients in 2019 and will investigate the role of early SBRT (50–60 Gy) to the primary tumor.
CONCLUSIONS
The recent advances in EGFR M+ NSCLC and the availability of different treatment options enables physicians to follow a patient-centric treatment approach. Osimertinib is now an option in the first-line setting, and approximately 1 in 4 patients will remain on treatment after 3 years. Approximately 50% of NSCLC patients will, however, require second-line therapy after about 1.5 years with osimertinib; for the majority of cases in the FLAURA trial, cytotoxic chemotherapy was the second-line treatment. In our opinion, cytotoxic chemotherapy in the second-line setting will most probably be replaced by the quadruple chemoimmunotherapy regimen of the IMPOWER150 trial. The observed plateau in the survival curves in the NSCLC subpopulation with activating mutations together with the aforementioned benefits are rational arguments that would justify this approach.
If one of the most import treatment goals for NSCLC patients is to delay giving cytotoxic chemotherapy for as long as possible, a second-generation EGFR (e.g., afatinib) followed by osimertinib with a median treatment duration of approximately 30 months might be an alternative option. Interestingly, the treatment duration of osimertinib as second- or later-line therapy was 20.2 months in the pooled analysis of the LUX-LUNG 3, 6 and 7 trials, which is almost identical to the treatment duration when given in the first-line setting. The AURA study highlights that activity of osimertinib in patients with tumors negative for T790M is less favorable than for EGFR T790M+ patients, and chemotherapy seems to be a better choice for these patients. It is likely that question of the best sequence can only be answered, when different EGFR mutations are seen as distinct diseases and additional biomarkers (e.g., c-MET overexpression, co-mutations, loss of EGFR T790M at progression) will be implemented into clinical decision making.
TAKE-HOME MESSAGES
-
Optimizing the treatment sequence of first-, second- and third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) is important to maximize benefits and prolong resistance for patients with EGFR mutant-positive non-small cell lung cancer (NSCLC).
-
Immunotherapy combination regimens, as demonstrated in the IMPOWER150 trial, are fast emerging as a second- or subsequent-line treatment option for EGFR M+ NSCLC patients.
-
Targeted therapy based on individual EGFR mutation profiles (EGFR mutations & co-mutations) has the potential to further improve outcomes and be more cost-effective.
COI
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a
potential conflict of interest.
Author Contributions
All authors contributed to and approved the final manuscript.