Introduction
Oxidized phospholipids
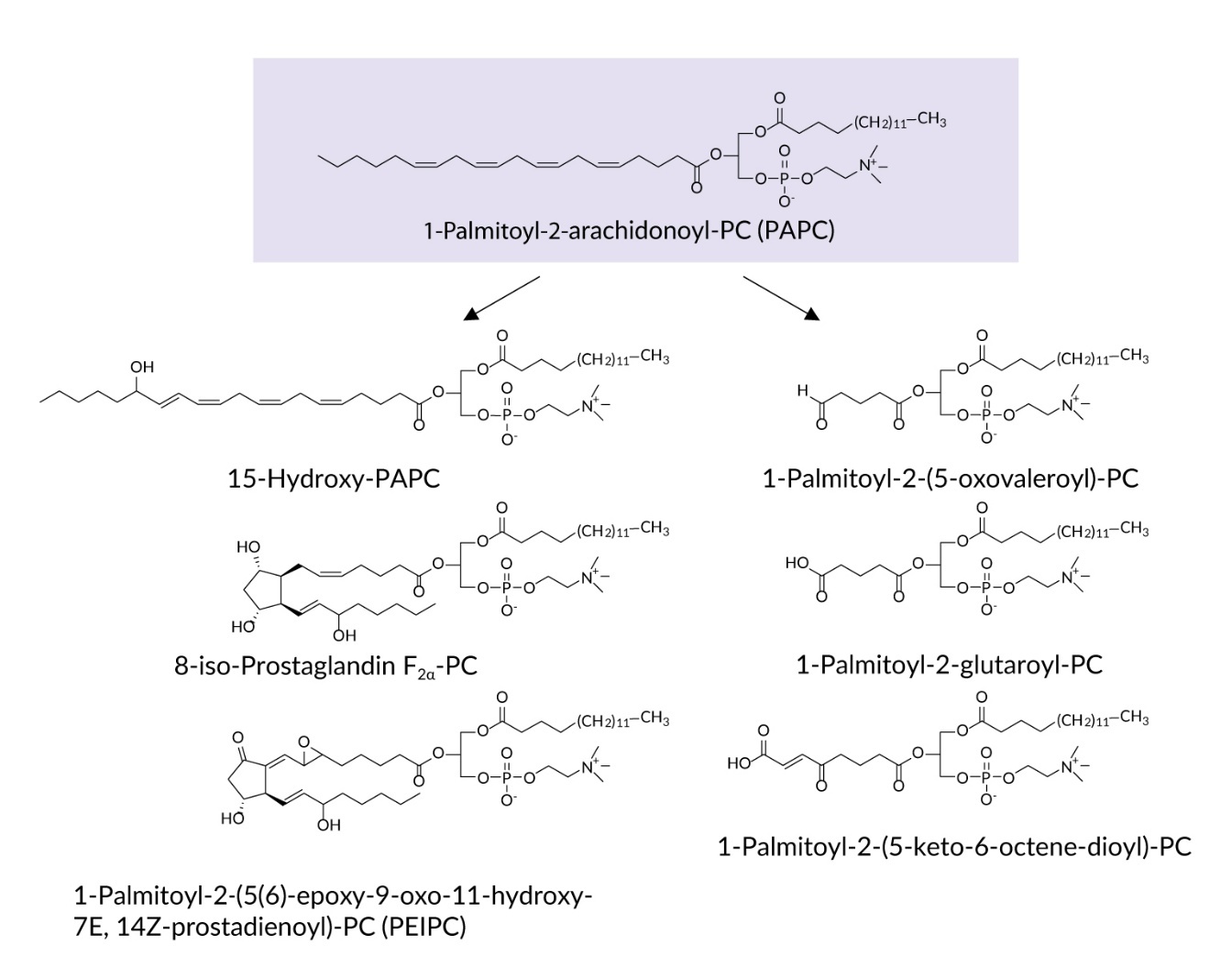
Tumor cells are characterized by oxidative stress resulting from multiple endogenous mechanisms such as rapid cellular proliferation and increased metabolism, as well as exogenous factors such as chemo- or radiotherapy.1 Overproduction of reactive oxygen species (ROS) is intimately involved in all facets of cancer biology including induction of oncogenic mutations, enhanced proliferation and reduced apoptosis, as well as neovascularization, invasion and metastasis.2 In this review, we discuss evidence suggesting that some effects of oxidative stress in tumors may be mediated by a family of lipid peroxidation products called oxidized phospholipids (OxPLs, Figure 1).
.jpeg)
OxPLs are increasingly recognized as biologically active lipids that are generated either as a result of oxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids, or by re-esterification of oxidized fatty acids (often called oxylipins) to the phospholipid scaffold via the enzymatic Lands’ cycle.3 Many oxylipins are biologically active in free, non-esterified form, for example prostaglandins and leukotrienes that are generated by cyclooxygenase (COX) and lipoxygenase (LOX) enzymes. It has been hypothesized that recycling of oxylipins to PLs and repeated PLA2-dependent release can have significant influence on time kinetics and magnitude of biological effects after repeated cycles of esterification and release.4 OxPLs are elevated in cardiovascular and other diseases5; experimental evidence in animal models suggests that OxPLs act as causative pathogenic factors in a variety of diseases such as atherosclerosis,6 liver fibrosis,7 nonalcoholic fatty liver disease,8 osteoporosis9 and ischemia-reperfusion injury.10 This review summarizes accumulating evidence on the involvement of OxPLs in tumor biology.
Accumulation of OxPLs in tumors
Available data show that human tumors often contain elevated levels of OxPLs.11–13 In addition to enhanced production of ROS and impaired antioxidant capacity of tumor and stromal cells, OxPLs accumulate in tumors due to the abundance of apoptotic and necrotic cells that are known to contain high amounts of oxidized phosphatidylcholine (OxPC).14 In addition to human samples, elevation of intratumor OxPLs was also documented in animal models. For example, elevated levels of OxPC containing aldehyde and carboxyl groups were detected in an animal model of familial adenomatous polyposis starting at an early phase of intestinal polyp formation.15 Accumulation of OxPC has been shown by immunohistochemical analysis of implanted melanoma and colon adenocarcinoma cell lines.16
Causative link between the accumulation of OxPLs and tumorigenesis is supported by the data showing that mice with genetically modified levels of peroxiredoxin 6 (Prdx6) in keratinocytes had changed susceptibility to skin carcinogenesis.17 Prdx6 is the only member of the peroxiredoxin family that has been reported to reduce OxPLs.18 Knockout of this protein in mouse skin keratinocytes resulted in elevated OxPLs levels and increased tumorigenesis, while the opposite effect was observed in transgenic animals overexpressing Prdx6.17 In addition to tumorigenesis susceptibility, Prdx6 also influenced progression of skin tumors in these animals. These data point to functional involvement of OxPLs in tumor initiation and progression. Phospholipidomic analysis documented the presence of increased concentrations of OxPCs not only in tumor tissues but also in plasma and bile of patients with non-small cell lung cancer, breast cancer and cholangiocarcinoma thus pointing to potential role of these lipids as biomarkers of malignant disease.11,13,19,20
In summary, available data documented accumulation of OxPLs in different types of tumors, their likely involvement in tumorigenesis, as well as potential role as biomarkers. Mechanisms, whereby OxPLs modulate tumor initiation and progression are discussed in the following paragraphs.
Biological effects of OxPLs in tumor cells
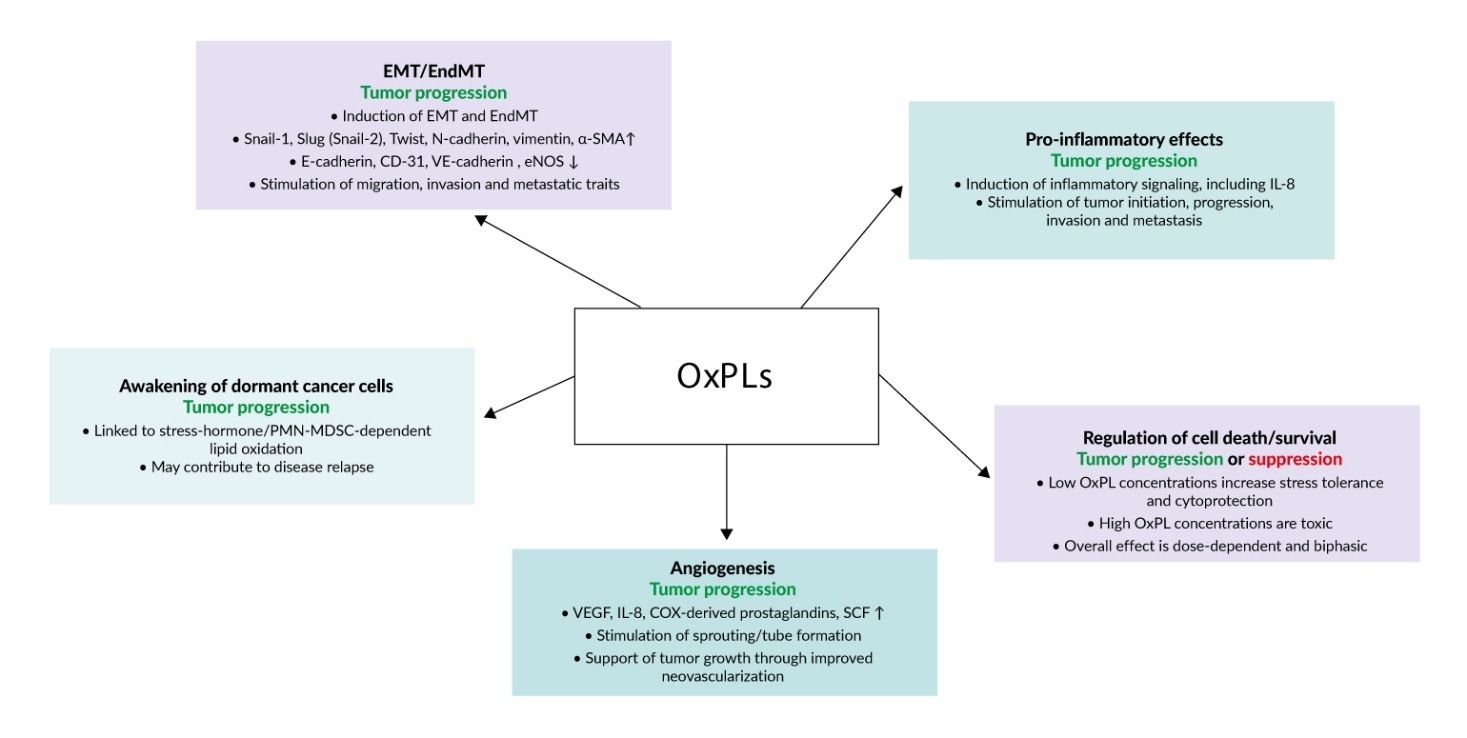
OxPLs exert pleiotropic effects on tumor biology, including the promotion of epithelial–mesenchymal transition (EMT), angiogenesis, reawakening of dormant cancer cells, cytoprotective responses and proinflammatory signaling. These diverse biological activities are summarized in Figure 2.
_on_tumor_cells.jpeg)
Epithelial-to-mesenchymal transition
EMT is a hallmark of oncogenesis characterized by loss of adhesion structures such as adherens junctions, tight junctions and desmosomes, and enhanced expression of α-smooth muscle actin, fibronectin, vimentin and other proteins necessary for migration and invasion. As a result of EMT, tumor cells lose their epithelial morphology and acquire mesenchymal traits that allow the cells to invade, migrate and adapt to the aggressive conditions of the intratumor milieu.21 OxPLs have been shown to stimulate EMT in several histologically different tumor cell lines.12 Treatment of cells with individual molecular species of OxPLs such as 1-palmitoyl-2-(5’-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC), 1-(palmitoyl)-2-(5-keto-6-octene-dioyl) phosphatidylcholine (KOdiA-PC) or 1-palmitoyl-2-glutaroyl-sn-glycero-phosphocholine (PGPC) (Figure 1) upregulated EMT-driving transcription factors Snail-1, Slug (Snail-2) and Twist1/2, reduced expression of epithelial markers (E-cadherin) and increased levels of mesenchymal markers (N-cadherin and vimentin). Phenotypic modulation of cells was accompanied by stimulated autophagy flux, which played a key role in EMT and increased cell migration and invasion capabilities.12 Thus, OxPLs induce a switch to the mesenchymal phenotype and enhance metastatic properties of malignant cells.
Similarly to epithelial cells, phenotypic switch to the mesenchymal phenotype may develop in endothelial cells. Endothelial-to-mesenchymal transition (EndMT) is driven by similar signaling mechanisms as EMT (Snail-1, Slug (Snail-2) and Twist-1/2) and leads to the loss of characteristic endothelial markers PECAM-1/CD-31, VE-cadherin, vascular endothelial growth factor receptor 2 (VEGFR2) and endothelial nitric oxide synthase (eNOS). Similarly to EMT, EndMT upregulates α-smooth muscle actin, vimentin, fibronectin and other proteins associated with cellular motility.22 EndMT is an important event in solid tumors where endothelial cells become a source of stromal fibroblast-like cells supporting tumor growth.23 Furthermore, EndMT promotes angiogenesis, invasion, metastatic dissemination and resistance to anti-tumor treatment.24 It was shown that OxPLs promote EndMT. POVPC has been shown to induce EndMT as evidenced by characteristic cell shape change, reduction of endothelial marker CD31, as well as elevation of mesenchymal markers α-smooth muscle actin and vimentin. The transition was dependent on the activation of classical signaling pathways leading to EndMT such as Smad2/3, Slug (Snail-2) and Twist-1.25 In summary, induction of EndMT is likely to be an additional mechanism whereby OxPLs promote tumor progression.
Cancer cells in a mesenchymal state are known to be susceptible to ferroptosis, a type of cell death where oxidation of membrane PLs plays a key role. Enhanced sensitivity to ferroptosis is mediated by several mechanisms. Cells in mesenchymal state are characterized by elevated expression of zinc finger E-box binding homeobox 1 (ZEB1) which is a lipogenic factor.26 As a result of ZEB1 activation, incorporation of PUFAs in cancer cells is elevated thus increasing susceptibility to ferroptosis.27 Another example is upregulation of discoidin domain receptor tyrosine kinase 2 (DDR2) by EMT leading to induction of pro-ferroptotic proteins CHAC1 and PTGS2 and increased sensitivity of recurrent breast tumor cells to erastin-induced ferroptosis.28 In addition, cluster of differentiation 44 (CD44) promotes iron uptake in mesenchymal cells, making them vulnerable to ferroptosis.29 In summary, many cancer cell types are characterized by increased sensitivity to phospholipid oxidation and ferroptosis, which may be used for antitumor therapy.
Angiogenesis
Apart from induction of phenotypic transition of epithelial and endothelial cells, OxPLs stimulate another cellular phenomenon that is of high relevance for tumor biology, namely angiogenesis. Without neovascularization, tumors are limited in their growth. As discussed above, OxPLs can induce EndMT. According to classical paradigm, EndMT inhibits functional activities specific for endothelial cells, including pro-angiogenic tube formation.22 Paradoxically, partial EndMT seems to be obligatory for angiogenesis, because transcription factor Slug (Snail-2), which is a marker and a key driver of EndMT, is critically important for sprouting angiogenesis.30 Thus, the ability of OxPLs to induce EndMT does not contradict their known capacity to stimulate angiogenesis. It was shown that OxPLs induce several pro-angiogenic effects. OxPLs stimulated production of angiogenic mediators such as VEGF, interleukin (IL)-8, COX-derived prostaglandins31 and stem cell factor.32 Pro-angiogenic activity of OxPLs has been demonstrated in in vitro, ex vivo and in vivo models such as tube formation, spheroid sprout formation, aortic ring outgrowth assay and Matrigel plug model.31,33,34 The angiogenic effect of OxPLs critically depended on oxidation of a PUFA residue but was independent of the type of polar head group because different classes of phospholipids containing identical oxidized residues but different polar head groups (PC, PG, PA or PS) induced comparable upregulation of VEGF.31 Mixtures of oxidized molecular species such as OxPAPC,31 as well as individual molecular species such as POVPC, PGPC, hydroxides and hydroperoxides of palmitoyl-linoleoyl-phosphatidylcholine induced pro-angiogenic effects.31,33 In addition, ethanolamine phospholipids containing 2-ω-carboxyethylpyrrole groups that were generated by covalent interaction of oxidized products of docosahexaenoic acid with the amino group in the polar head of phospholipid demonstrated angiogenic activity.34
Several publications addressed signaling mechanisms of angiogenic activity of OxPLs. The data suggest that angiogenic effects of OxPLs are mediated by several signal transduction pathways including unfolded protein response,35,36 transcription factor NRF2,32,37 microRNAs (miR-66335 and miR-15532), as well as protein kinase CK2.32 In summary, available data support the notion that OxPLs stimulate angiogenesis and thus can promote tumor neovascularization and growth.
Awakening of dormant cancer cells
An interesting biological effect of OxPLs related to tumor biology is their ability to reactivate dormant cancer cells. Dormancy describes malignant cells that do not proliferate but also do not die. Such cells are resistant to therapy and represent a major source of disease relapses that may develop years after apparently successful therapy.38 Dormant cancer cells can be awakened by a variety of factors including inflammation, changes in extracellular matrix, action of angiogenic mediators, stimulation with cytokines such as transforming growth factors (TGFs), as well as other endo- and exogenous factors.39 Available data show that OxPLs can activate dormant cells in the presence of stress hormones and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC). It was found that stimulation of β2-adrenoreceptors in PMN-MDSC resulted in secretion of S100A8/A9 proteins, activation of myeloperoxidase and oxidation of lipids, which in turn stimulated proliferation of dormant cancer cells.40 Plasmalogen-PE-adduct with 4-hydroxynonenal and lyso-PE were identified as molecular species responsible for the awakening activity. Thus, activation of hibernating malignant cells is likely to be an additional mechanism whereby OxPLs can stimulate tumor progression.
Cell-protective action of low OxPL concentrations
In full agreement with the Paracelsus’ maxim “the dose makes the poison”, OxPLs induce a biphasic effect on cell survival. While high concentrations of OxPLs are toxic and selectively kill tumor cells,41,42 at low concentrations OxPLs are not toxic and can protect macro- and microvascular endothelial cells from cytotoxic action of serum deprivation and cytostatics such as staurosporine, camptothecin, arabinoside C and doxorubicin.43 Such protective effects may play a role in resistance of tumors to cytostatics.
Pro-inflammatory activity of OxPLs
Inflammation is known to promote all stages of tumor development from initiating stages to invasion and metastasis.44 Inflammation is a promising but essentially unexplored field linking OxPLs and tumor biology. Available data are consistent with the notion that OxPLs may actively support tumor progression through their well described pro-inflammatory activity. The data discussed in previous paragraphs document the presence of elevated levels of OxPLs in tumor tissues. On the other hand, a large body of data characterize OxPLs as active pro-inflammatory molecules.45 OxPLs are known to induce IL-8,46 which is well known for its role in tumor progression.47 Thus, OxPL-induced inflammation is likely to represent an additional mechanism coupling oxidative stress with tumor initiation and progression.
Oxidized phospholipids in tumor ferroptosis
Oxidation of membrane phospholipids plays a key mechanistic role in ferroptosis, which is a form of cell death driven by massive intracellular lipid peroxidation.48 It differs from other types of cell death, like apoptosis, autophagy, anoikis, necroptosis, or pyroptosis, which are regulated by enzymatic cascades of protein kinases and proteases.49 Compared to other cell death types, ferroptosis is mediated by toxic peroxidized lipids but not proteins.48 It has been shown that ferroptosis is accompanied by predominant oxidation of phospholipids and especially phosphatidylethanolamine containing doubly- and triply-oxygenated species of arachidonic and adrenic fatty acids, which strongly correlated with the ferroptotic process.50,51 The critical importance of phospholipid oxidation in ferroptosis is strongly supported by protection of cells upon inhibition of enzymes synthesizing peroxidation-prone polyunsaturated phospholipids.52 Furthermore, ferroptosis is triggered by inhibitors of GPX4 and inhibited by overexpression of GPX4 – the only isoform of the GPX enzymes capable of reducing peroxidized fatty residues within phospholipid molecules, while other members of the family accept as substrates only free, non-esterified fatty acids.
Malignant transformation often changes sensitivity of cells to ferroptosis.42 This can be due to mutations in oncogenes or tumor suppressor genes and, as described below, can significantly influence sensitivity of tumors to anti-tumor immunity and therapeutic ferroptosis.
Regulation of tumor cell ferroptosis by tumor suppressor and oncogene proteins
Many tumor suppressors and oncogenes regulate expression or activity of enzymes controlling intracellular lipid peroxidation. Tables 1 and 2 describe the mechanisms underlying changed sensitivity of tumor cells to ferroptosis.
These data show that the relationship between oncogenic transformation of cells and their sensitivity to phospholipid oxidation is not straightforward. Sensitivity of tumor cells to ferroptosis can be changed both by inactivation of tumor suppressors as well as uncontrolled activation of oncogenes. Importantly, regulation of ferroptosis by oncogenic mutations occurs in both directions, i.e., some mutations increase sensitivity of tumors to ferroptosis, while others induce the opposite effect. Modulation of sensitivity to phospholipid oxidation and ferroptosis by tumor suppressors and oncogenes is mediated by changed expression or activities of proteins that exert different functions in lipid peroxidation including those regulating iron handling (transferrin receptor, NCOA4), enzymes synthesizing peroxidation-prone PUFA-phospholipids (ACSL4), antioxidant enzymes (GPX4) and proteins involved in production of antioxidant proteins (NRF2) or small antioxidant molecules (SLC7A11, FSP1). Thus, it is tempting to speculate that identification of driving mutations of individual tumors and their effects on lipid peroxidation can guide personalized ferroptosis-based therapy.
Influence of metabolic factors on ferroptosis sensitivity
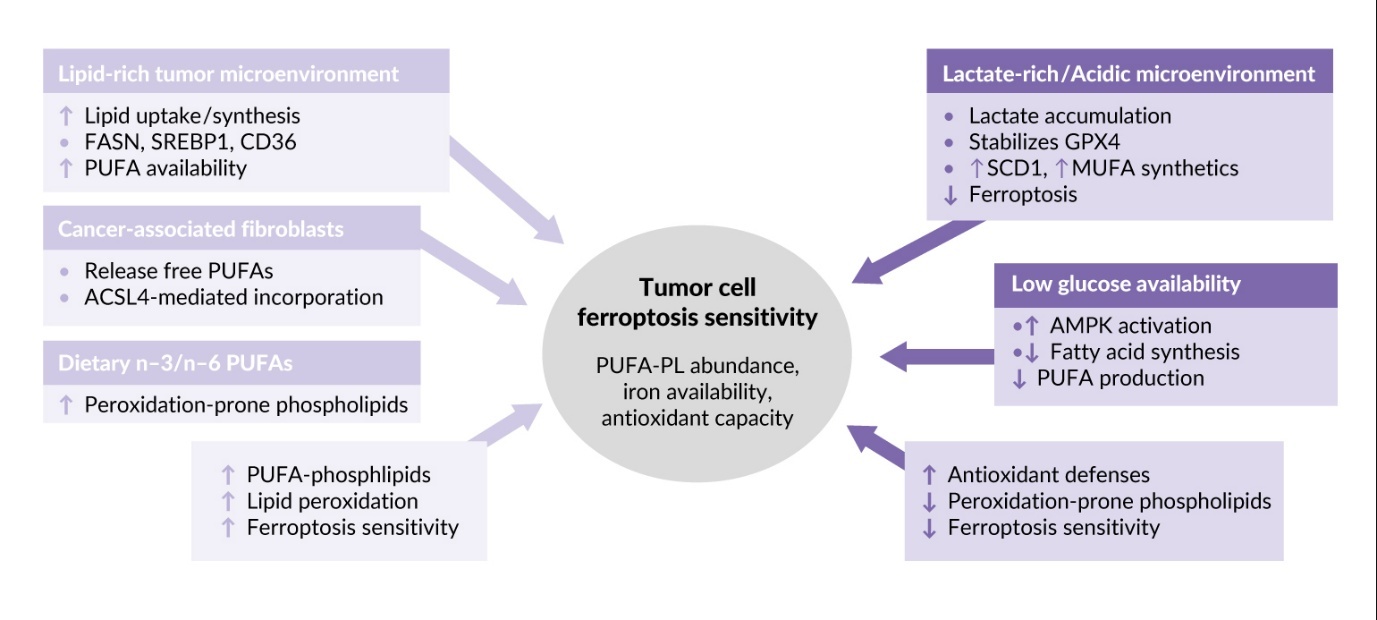
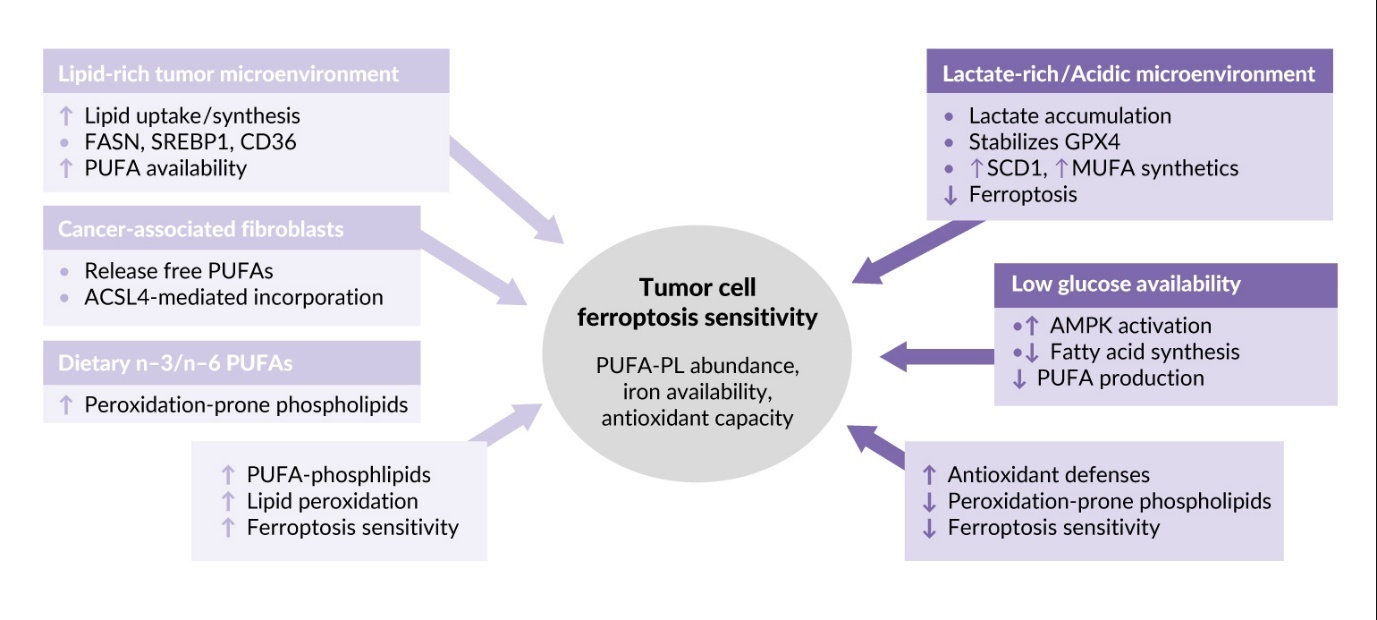
In addition to the type of oncogenic mutation (Tables 1 and 2), the sensitivity of tumor cells to phospholipid oxidation and ferroptosis is regulated by the tumor microenvironment (TME). This term describes a combination of factors inside and around the tumor, such as the presence of stromal and immune cells, blood capillaries, extracellular matrix and extracellular fluid. The TME is typically characterized by hypoxia, acidosis and nutrient deprivation. Effects of characteristic metabolic abnormalities of TME on tumor cell sensitivity to lipid peroxidation and ferroptosis are described below and summarized in Figure 3.
Rapidly growing cancer cells require lipids as sources of energy and building blocks for membranes and therefore upregulate lipid synthesis and uptake through FASN, SREBP1 and CD36, ultimately leading to accumulation of lipids containing peroxidation-prone PUFAs.68–71 In addition, cancer associated fibroblasts participate in creating lipid-rich TME by secretion of free PUFAs that are incorporated into phospholipids of tumor cell membranes by ACSL4, which is highly expressed in many tumors, rendering them sensitive to ferroptosis.52,72 Thus, lipid-rich TME is a factor promoting ferroptosis in solid tumors. This property may be used in therapeutic purposes because dietary supplementation with n-3 and n-6 PUFAs leads to their incorporation into membranes instead of monounsaturated fatty acids (MUFAs), thus resulting in increased tumor ferroptosis.73
In contrast to lipid enrichment, which promotes peroxidation of lipids characteristic of ferroptosis, other TME components can inhibit this type of cell death. Ferroptosis is inhibited by lactate which is abundant in the TME due to active aerobic glycolysis typical of tumors.74,75 Furthermore, accumulation of lactate is promoted by active glutaminolysis, which is a critical metabolic pathway in many tumors76 producing lactate via malic and LDH enzymes.77 Lactate-induced acidification stabilizes GPX474 and upregulates stearoyl-coenzyme A (CoA) desaturase-1 leading to enhanced production of anti-ferroptotic MUFAs.78 Inhibition of ferroptosis can also result from low glucose availability in the TME, which triggers AMP-activated protein kinase (AMPK)-mediated phosphorylation of acetyl-CoA carboxylases and inhibits production of fatty acids, including PUFAs that are necessary for ferroptosis.79,80
To summarize, different TME elements play a context-dependent role in regulation of ferroptosis, which should be taken into account for developing personalized ferroptosis-based cancer therapy.
Importance of oxidized phospholipids and ferroptosis in antitumor immune response
The data discussed above support the notion that tumors differ from normal cells with respect to ferroptosis sensitivity, which is influenced by both the type of oncogenic mutation and factors present in the intratumor milieu. Susceptibility to ferroptosis can significantly influence proliferation of tumor cells and their resistance to the immune system. Below, two important phenomena are discussed, namely ferroptosis as a weapon of immune cells against tumors, as well as the opposing phenomenon, i.e., ferroptosis as a mechanism suppressing immune defense.
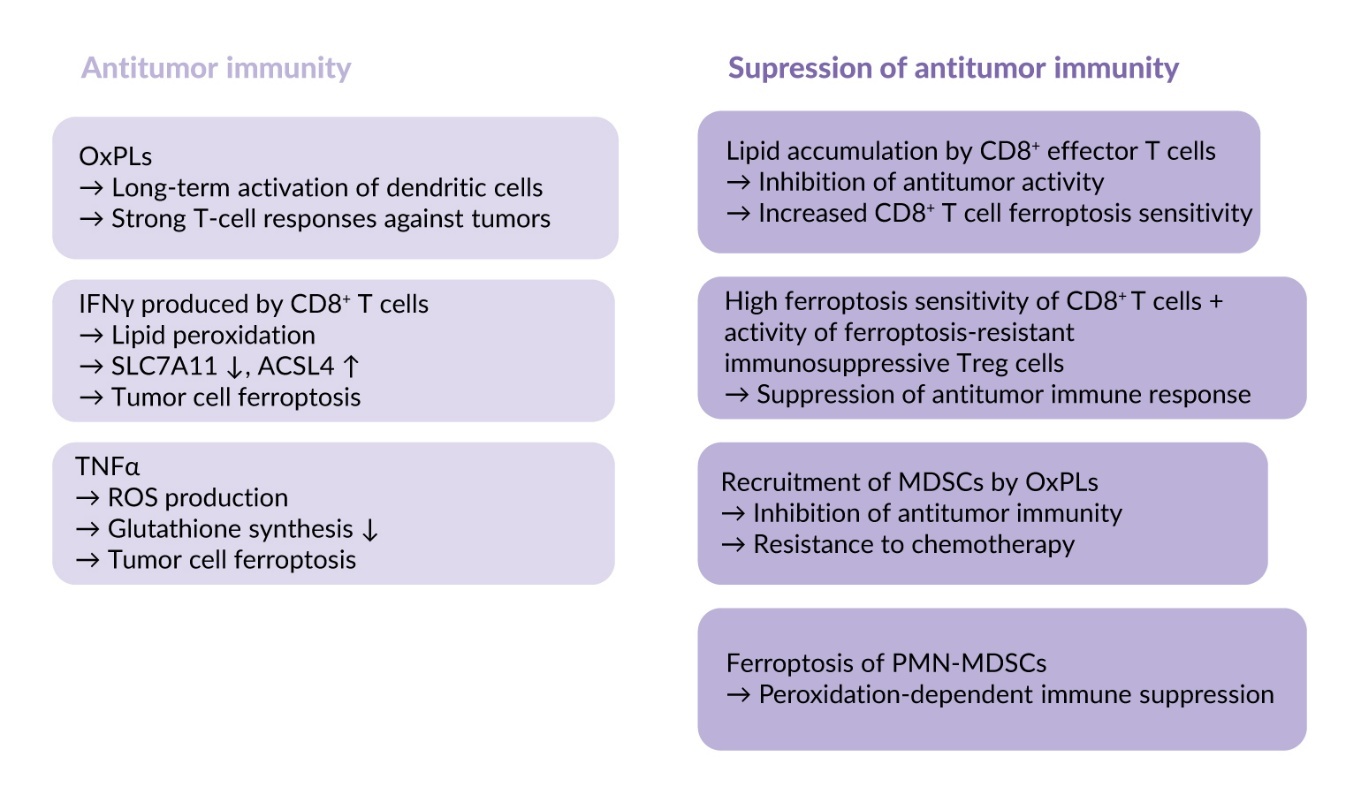
Oxidized phospholipids and ferroptosis as mechanisms of antitumor immunity
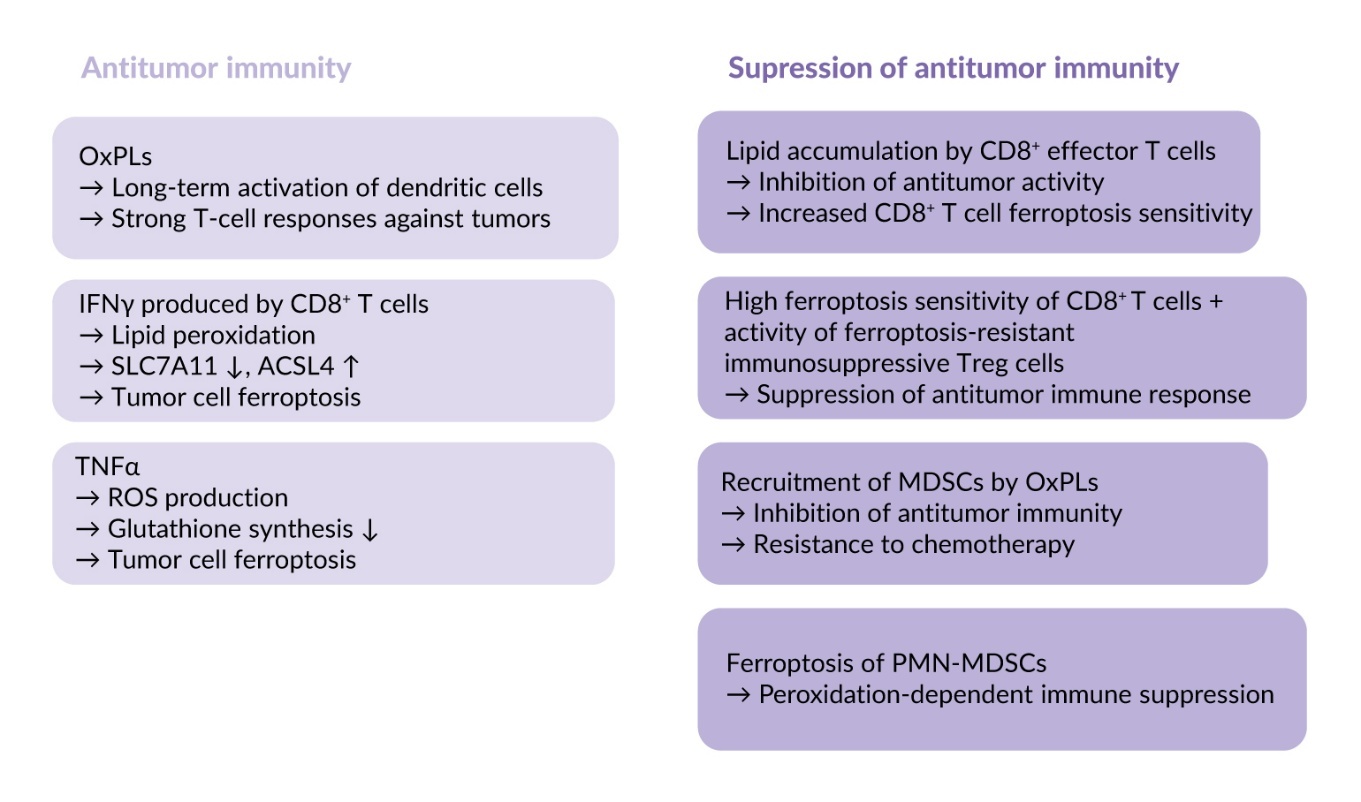
OxPLs such as OxPAPC and PGPC can induce long-term activation of dendritic cells, the so-called “hyperactivation state.”81 Extracellular protein CD14 binds OxPLs and translocates them into dendritic cells thus leading to inflammasome activation and production of large amounts of interleukin-1β, known as an active regulator of differentiation of T cells and their effector functions.81 These hyperactivated dendritic cells were capable of inducing strong T-cell responses against tumor lysates in vitro and tumors in vivo.82 Thus, OxPLs can directly stimulate antitumor immunity through their effects on dendritic cells.
Another antitumor mechanism activated by phospholipid oxidation depends on the action of cytokines produced by intratumor immune cells. These cytokines can kill malignant cells by inducing ferroptosis. Interferon gamma (IFNγ) produced by CD8⁺ T cells activates lipid peroxidation and promotes ferroptosis in different tumors, such as colon adenocarcinoma, melanoma and breast cancer, in murine and human models. The underlying mechanisms are downregulation of SLC7A11 which reduces glutathione production, as well as upregulation of ACSL4, which stimulates accumulation of PUFAs-containing phospholipids.83,84 Another cytokine that stimulates ferroptosis in tumors is transforming growth factor (TGF) β1, which promotes cellular peroxidation through SMAD transcription factors that downregulate SLC7A11 expression in liver cancer cells.85 In parallel, tumor necrosis factor alpha (TNFα) stimulates ROS production and inhibits glutathione synthesis, thus sensitizing tumors to ferroptosis.86 Thus, ferroptosis is an efficient effector mechanism of the immune system mediating tumor cell elimination.
Role of ferroptosis in suppression of anti-tumor immunity
Data summarized in the previous paragraph show that the immune system induces antitumor effects by enhancing sensitivity of tumor cells to ferroptosis. However, oxidation of phospholipids and ferroptosis can negatively influence activity of immune cells. It has been shown that upon homing to the tumor bed CD8⁺ T cells accumulate lipids via CD36-mediated lipid uptake, which lowers antitumor properties and makes them prone to ferroptosis.16,87 Other types of T cells are also sensitive to ferroptosis, such as follicular helper T cells that are important for B-cell activation.88 In contrast to effector T cells, regulatory T cells (Tregs), which inhibit immune responses by dampening effector T-cell activation, are ferroptosis-resistant and therefore able to preserve their immunosuppressive properties under conditions promoting ferroptosis.89–91 As a net effect, the combination of high sensitivity of CD8⁺ effector T cells with resistance of immunosuppressive Treg cells leads to suppression of anti-tumor immune response.
In addition to the immunosuppressive mechanisms described above, OxPLs have been shown to recruit myeloid-derived suppressor cells (MDSCs) that inhibit antitumor immunity and mediate resistance to chemotherapy. It has been shown that treatment of mouse lung cancer model with cisplatin and other cytostatics induced accumulation of OxPAPC, recruited MDSCs and promoted tumor growth.92 Injection of OxPAPC induced accumulation of MDSC in vivo thus supporting the involvement of OxPLs in mobilization of these immunosuppressive cells.
An additional peroxidation-dependent mechanism of immune suppression in tumors results from ferroptosis of MDSCs generated from neutrophils (PMN-MDSCs), which are well-recognized inhibitors of antitumor immune responses. Spontaneous intratumor ferroptotic death of these cells leads to release of peroxidized lipids, including oxidized phosphatidylethanolamine, resulting in reduced proliferation and dysfunction of T cells with corresponding immune suppression.93
In summary, oxidation of phospholipids is a crucial step in ferroptosis that represents an important effector mechanism of antitumor immunity; however, it also induces multiple immune-suppressive effects (summarized in Figure 4). This dual activity represents an important challenge for developing an efficient anti-cancer therapy based on induction of ferroptosis.94,95
Conclusions
Previous studies of OxPLs have shown their important role in multiple pathologies including atherosclerosis, neurodegenerative diseases and other conditions.96 Rapidly accumulating data discussed above suggest that OxPLs are also active players in tumor generation and growth. OxPLs induce multiple effects in tumor progression, which further characterizes them as pleiotropic lipid mediators. OxPLs were shown to accumulate in the tumor milieu and influence not only tumor progression but also antitumor immune responses. An important conclusion from available data is that OxPLs often induce oppositely directed functional effects. This functional complexity apparently explains controversial therapeutic effectiveness of ferroptosis inducers, which was discussed in recent reviews.94,95 The data suggest that a better understanding of the mechanistic role of OxPLs in tumor biology is a prerequisite for improved efficiency of antitumor therapy.
A major limitation of the current literature is that the majority of evidence regarding the biological effects of OxPLs in tumors comes from in vitro systems and animal models, with only limited validation in human clinical samples. While these experimental approaches have been invaluable for identifying potential mechanisms, they may not fully capture the complexity, heterogeneity and temporal dynamics of the human TME. As a result, the clinical relevance of many reported findings remains uncertain, necessitating more standardized translational studies and well-annotated patient cohorts to define the precise role of OxPLs in human cancer.
Conflict of interest
The authors declared that the manuscript was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors declared that no financial support was received from any organization for the submitted work.
Author contributions
All authors contributed to and approved the final manuscript.
AI use
No artificial intelligence (AI) or generative AI tools were used in the preparation of this manuscript.