Introduction
Multiple myeloma (MM) accounts for approximately 10% of the hematologic malignancies and 1% of all cancers.1–4 While most MM patients eventually relapse and develop treatment resistance, substantial progress over the past 20 years has translated into deeper, more durable remissions and prolonged survival.3,5,6 The introduction of high-dose therapy (HDT) followed by autologous stem cell transplantation (ASCT) transformed the treatment landscape of newly diagnosed MM (NDMM) for eligible patients. More recent advances include proteasome inhibitors (PIs) such as bortezomib, carfilzomib and ixazomib,7 immunomodulatory drugs (IMiDs) such as lenalidomide, thalidomide and pomalidomide,8 and anti-CD38 monoclonal antibodies (mAbs) such as daratumumab and isatuximab.9 Collectively, these agents form the backbone of contemporary combination regimens. In addition, new therapeutic modalities, including antibody-drug conjugates (ADCs), bispecific T-cell engagers and chimeric antigen receptor (CAR) T cells, are being evaluated and increasingly integrated earlier in the treatment paradigm.5,6,10–12
The treatment algorithm for NDMM varies according to frailty, age, comorbidities and cytogenetic risk.13 For transplant-eligible (TE) patients, the recommended regimen is induction therapy, typically including at least a triplet drug combination of an IMiD, PI and corticosteroid, with quadruplet regimens incorporating an anti-CD38 mAb, followed by HDT/ASCT and lenalidomide maintenance. A commonly used regimen is a combination of daratumumab, bortezomib, thalidomide and dexamethasone (D-VTd). The European Hematology Association and European Society for Medical Oncology (EHA-ESMO) guidelines list D-VTd as a standard of care (SoC) for TE patients,13 while the US National Comprehensive Cancer Network (NCCN) guidelines recommend replacing thalidomide with lenalidomide (D-RVd) and also include isatuximab-based regimen comprising isatuximab, lenalidomide, bortezomib and dexamethasone (Isa-RVd).14 For transplant-ineligible (TIE) patients, standard options include daratumumab plus bortezomib, melphalan and prednisone (D-VMP), daratumumab, lenalidomide and dexamethasone (D-Rd), D-RVd and RVd, with dose adjustments guided by frailty and tolerability.13,14
This review discusses several interconnected paradigm shifts that define the current management of NDMM, with emphasis on recent trial updates and their implications for clinical practice. These include the evolution from triplet to quadruplet therapy incorporating anti-CD38 mAbs and the early integration of bispecific antibodies, ADCs and next-generation cereblon E3 ligase modulatory drugs (CELMoDs) into treatment protocols. We also review the emerging role of minimal residual disease (MRD) negativity and genetic determinants in guiding treatment decisions, particularly regarding the selective use of high-dose melphalan and ASCT in NDMM. Table 1 summarizes the key clinical trials discussed in this review.
From Triplet to Quadruplet Therapy: Anti-CD38 Antibodies
Daratumumab-based regimens: GRIFFIN and CASSIOPEIA in TE NDMM
Daratumumab, the first-in-class CD38-targeting mAb, has demonstrated significant clinical efficacy in relapsed or refractory (RR) MM,53–57 both as a monotherapy and in combination with standard regimens. In NDMM, daratumumab-based regimens have improved the depth and durability of response and progression-free survival (PFS) in both TIE and TE settings.15,16,19,58,59
The large randomized phase II GRIFFIN trial which also incorporated a safety run-in cohort of 16 patients was the first study to evaluate the addition of daratumumab to the RVd backbone in TE NDMM.15,60 Patients (n=207) were randomized 1:1 to receive D-RVd or RVd induction, followed by ASCT, D-RVd or RVd consolidation and R-D or lenalidomide maintenance. The primary endpoint, stringent complete response (sCR) rate by the end of post-ASCT consolidation, favored D-RVd versus RVd (42.4% vs 32.0%; odds ratio: 1.57 [95% CI: 0.87–2.82]; 1-sided p=0.068), with responses deepening and MRD negativity rates improving over time.15 With extended follow-up, the D-RVd arm demonstrated sustained PFS benefit, including a 4-year PFS rate of 87.2% versus 70.0% for RVd (HR: 0.45 [95% CI: 0.21–0.95]; p=0.032).16 Adding daratumumab to RVd improved outcomes in key subgroups, including patients with high-risk cytogenetic abnormalities, and yielded meaningful quality-of-life (QoL) benefits.17,18 These findings positioned D-RVd as a core quadruplet strategy in TE NDMM and were subsequently validated in the phase III PERSEUS trial, which demonstrated significantly higher rates of deep response and MRD negativity, as well as improved PFS versus RVd,19 providing the basis for NCCN guideline endorsement and regulatory approval of the regimen by the US Food and Drug Administration (FDA).
Earlier phase III data from CASSIOPEIA established D-VTd as an effective daratumumab-based quadruplet for TE NDMM, and updated analyses showed sustained PFS benefit with daratumumab maintenance, including among patients who had received daratumumab during induction and consolidation.20,21 Together, these findings provided both context and the foundation for the recent daratumumab-based studies summarized below, including in TIE/transplant-deferred, post-ASCT and frail populations.
CEPHEUS: D-RVd in TIE or transplant-deferred NDMM
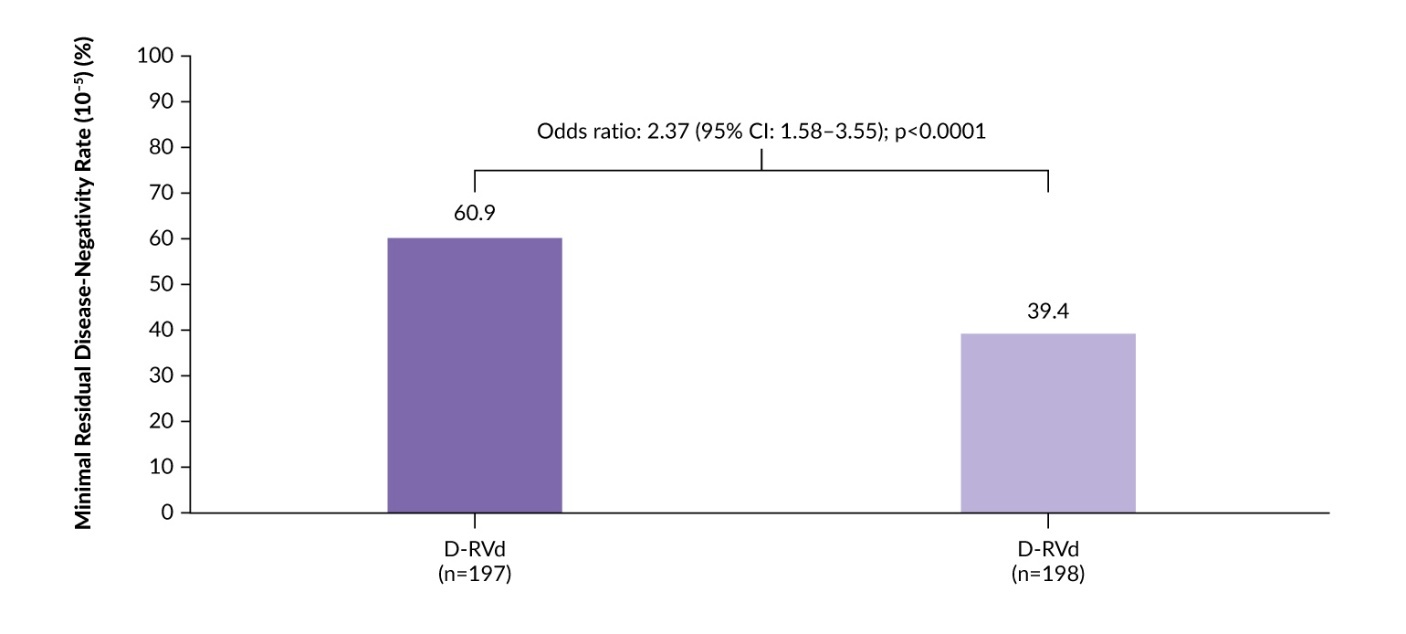
The phase III CEPHEUS trial evaluated subcutaneous daratumumab plus RVd in patients with NDMM who were TIE or had deferred transplant.22 A total of 395 patients were randomized 1:1 to eight cycles of D-RVd or RVd, followed by D-Rd or Rd until progression. The primary endpoint was MRD negativity at the 10−5 threshold. Key secondary endpoints included complete response or better (≥CR) rate, PFS and sustained MRD negativity.
The addition of daratumumab to RVd was associated with deeper and more durable responses.22,23 At a median follow-up of 58.7 months, the MRD negativity rate was 60.9% with D-RVd versus 39.4% with RVd alone (odds ratio: 2.37 [95% CI: 1.58–3.55]; p<0.0001) (Figure 1).22 The ≥CR rates were 81.2% versus 61.6% and sustained MRD negativity (≥12 months) rates were 48.7% versus 26.3%, respectively. Expanded analysis showed consistently higher cumulative MRD negativity with D-RVd at both 10−5 and 10−6 sensitivity across all prespecified timepoints, with nearly two-fold higher sustained MRD negativity (≥CR) rates at 12, 24 and 36 months.23
PFS was also significantly prolonged with D-RVd versus RVd (median, not reached vs 52.6 months), with 54-month PFS rates of 68.1% versus 49.5%, respectively (HR: 0.57 [95% CI: 0.41–0.79]; p=0.0005).22 PFS benefits were maintained regardless of MRD negativity status (at 10−6) and across prespecified subgroups.22,23 Overall survival (OS) data were immature but showed a numerical trend favoring D-RVd (HR: 0.85).22 The safety profile of the combination regimen was consistent with those previously reported for daratumumab and RVd. The most common grade 3–4 treatment-emergent adverse events (TEAEs) were neutropenia (44.2% with D-RVd vs 29.7% with RVd) and thrombocytopenia (28.4% vs 20.0%). In summary, CEPHEUS supports D-RVd as a new SoC for TIE or transplant-deferred patients with NDMM.
_negativity_at.jpeg)
GEM2017Fit: Daratumumab as part of quadruplet induction or Rd consolidation regimens in fit elderly patients
The phase III GEM2017Fit trial evaluated the addition of daratumumab to carfilzomib, lenalidomide and dexamethasone (D-KRd) versus standard triplet regimens in fit elderly patients with NDMM.24,25 A total of 462 patients aged 65–80 years were randomized 1:1:1 to 18 cycles of KRd, 18 cycles of D-KRd or nine cycles of VMP followed by Rd. Within the triplet cohorts, induction was followed by D-Rd consolidation; then patients were stratified by MRD status to D-R maintenance or observation. The primary endpoint was MRD negativity rate post-induction.
The trial met its primary endpoint, demonstrating significantly higher rates of MRD negativity (10−5) with D-KRd (61%, OR: 2.03 [95% CI: 1.61–2.57]; p<0.001) and KRd (54%, OR: 1.74 [95% CI: 1.39–2.16]; p<0.001) compared to VMP/Rd (27%).24 Subsequent D-Rd consolidation further improved MRD negativity rates across all arms. PFS was significantly prolonged with both D-KRd (HR: 0.66 [95% CI: 0.45–0.96]; p=0.032) and KRd followed by D-Rd consolidation (HR: 0.60 [95% CI: 0.40–0.88]; p=0.009) compared with VMP/Rd followed by D-Rd. At a median follow-up of 46 months, 50-month PFS rates were 67%, 71% and 52% with D-KRd, KRd and VMP/Rd, respectively. The incidence of treatment-related AEs during consolidation remained low. In summary, while D-Rd consolidation deepened responses, VMP/Rd remained inferior to KRd-based induction, with comparable outcomes observed when daratumumab was introduced during induction or consolidation.
AURIGA: Daratumumab plus lenalidomide maintenance post-ASCT
Maintenance therapy is a cornerstone of post-ASCT management. However, many patients remain MRD-positive after ASCT, representing a high-risk state. The phase III AURIGA study evaluated whether adding daratumumab to lenalidomide maintenance could deepen responses and improve outcomes.26 The trial enrolled post-transplant NDMM patients with a very good partial response or better (≥VGPR) who were MRD-positive (threshold 10–5) and naïve to CD38-targeting mAb. A total of 230 patients were randomized 1:1 to D-R (n=99) or R (n=101) maintenance for up to 36 cycles. The primary endpoint was the MRD-negative (10–5) conversion rate by next-generation sequencing (NGS) at 12 months.
At a median follow-up of 32.3 months, D-R significantly enhanced MRD-negative conversion: 50.5% versus 18.8% for R alone (OR: 4.51 [95% CI: 2.37–8.57]; p<0.0001).26 Moreover, D-R deepened responses at the 10−6 sensitivity and improved ≥CR rates (75.8% vs 61.4%). This translated into a significant survival benefit: the 30-month PFS rate was 82.7% with D-R versus 66.4% with R (HR: 0.53 [95% CI: 0.29–0.97]), representing a 47% reduction in the risk of progression or death.
A post hoc analysis demonstrated consistent benefits of D-R maintenance across all clinically relevant subgroups, including cytogenetically defined high-risk disease characterized by del(17p), t(4;14) or t(14;16) (OR: 6.53 [95% CI: 0.71–60.05]) and the standard-risk subgroup (OR: 4.64 [95% CI: 2.15–10.04]).27 PFS was consistently improved with D-R maintenance in both standard and high cytogenetic risk subgroups, regardless of the number of high-risk cytogenetic abnormalities, including patients with gain/amp(1q21). No unexpected safety concerns emerged. Together, these data support D-R maintenance in clinically relevant NDMM patient subgroups.
IFM2017-03: Corticosteroid-sparing D-R in frail patients
The phase III MAIA trial established D-Rd as a standard regimen for TIE NDMM, with sustained improvements in PFS and OS versus Rd.59 However, frail patients often experience suboptimal outcomes due to higher toxicity and treatment discontinuation rates, with prolonged corticosteroid exposure being particularly problematic in this population. The phase III IFM2017-03 trial was the first randomized study dedicated to frail patients, comparing D-R versus Rd with a corticosteroid-sparing design.28 A total of 295 patients aged ≥65 years with an Eastern Cooperative Oncology Group (ECOG)-based proxy frailty score ≥2 were randomized 2:1 to receive D-R (n=200) or Rd (n=95). The primary endpoint was PFS.
Median PFS was significantly longer in the D-R group at 53.4 months, compared with 22.5 months in the Rd group (HR: 0.51 [95% CI: 0.37–0.70]; p<0.0001).28 OS was also significantly improved: with the median not reached versus 47.2 months (HR: 0.52 [95% CI: 0.35–0.77]; p=0.0001), corresponding to a 48% reduction in the risk of death. Although grade ≥3 AEs were more frequent with D-R (89% vs 79%), the incidence of serious infections and pneumonia was comparable. Importantly, patients receiving D-R experienced meaningful improvements in QoL. These data establish the dexamethasone-sparing D-R regimen as a preferred option for frail patients.
Isatuximab-based regimens
Isatuximab, the second CD38-targeting mAb, has demonstrated efficacy in RRMM.61–64 In NDMM, the benefits of isatuximab plus RVd (Isa-RVd) were established in the phase III GMMG-HD7 and IMROZ trials. In GMMG-HD7, Isa-RVd induction improved MRD negativity in TE patients, with benefits deepening after ASCT.29,30 In IMROZ, Isa-RVd significantly improved CR and MRD negativity rates and prolonged PFS compared with RVd in TIE patients, leading to its regulatory approval in this setting.31 The phase III BENEFIT trial further addressed the contribution of PIs in TIE NDMM demonstrating that Isa-RVd significantly increased MRD endpoints compared with Isa-Rd, supporting bortezomib as an important component of anti-CD38-based therapy in TIE patients.32 Similarly, the phase III IsKia trial showed that adding isatuximab to KRd (Isa-KRd) increased MRD negativity rates in TE patients compared with the KRd triplet.33,34 High and sustained MRD negativity rates with Isa-KRd was also demonstrated in patients with high-risk NDMM, regardless of transplant eligibility, in the phase II GMMG-CONCEPT study.65,66 Recently published results of the MIDAS trial evaluating MRD-guided strategy following Isa-KRd induction are detailed below.
MIDAS: MRD-guided therapy with Isa-KRd
The phase III MIDAS trial represents an important development, not only for isatuximab but for the broader concept of MRD-guided treatment adaptation. With the introduction of novel induction regimens, it remains unclear whether these combinations are sufficiently potent to omit upfront ASCT or whether ASCT confers a clinically meaningful incremental benefit. While tandem ASCT within a 6-month interval is recommended in patients with high-risk disease, the benefit of ASCT appears limited in patients who achieve MRD negativity after induction.67,68 The concept of MRD-guided therapy was supported, for example, by the phase II MASTER trial, which demonstrated that MRD-guided consolidation and treatment cessation after D-KRd induction and ASCT could achieve MRD negativity in 81% of MRD-evaluable patients, with 71% subsequently discontinuing treatment.69
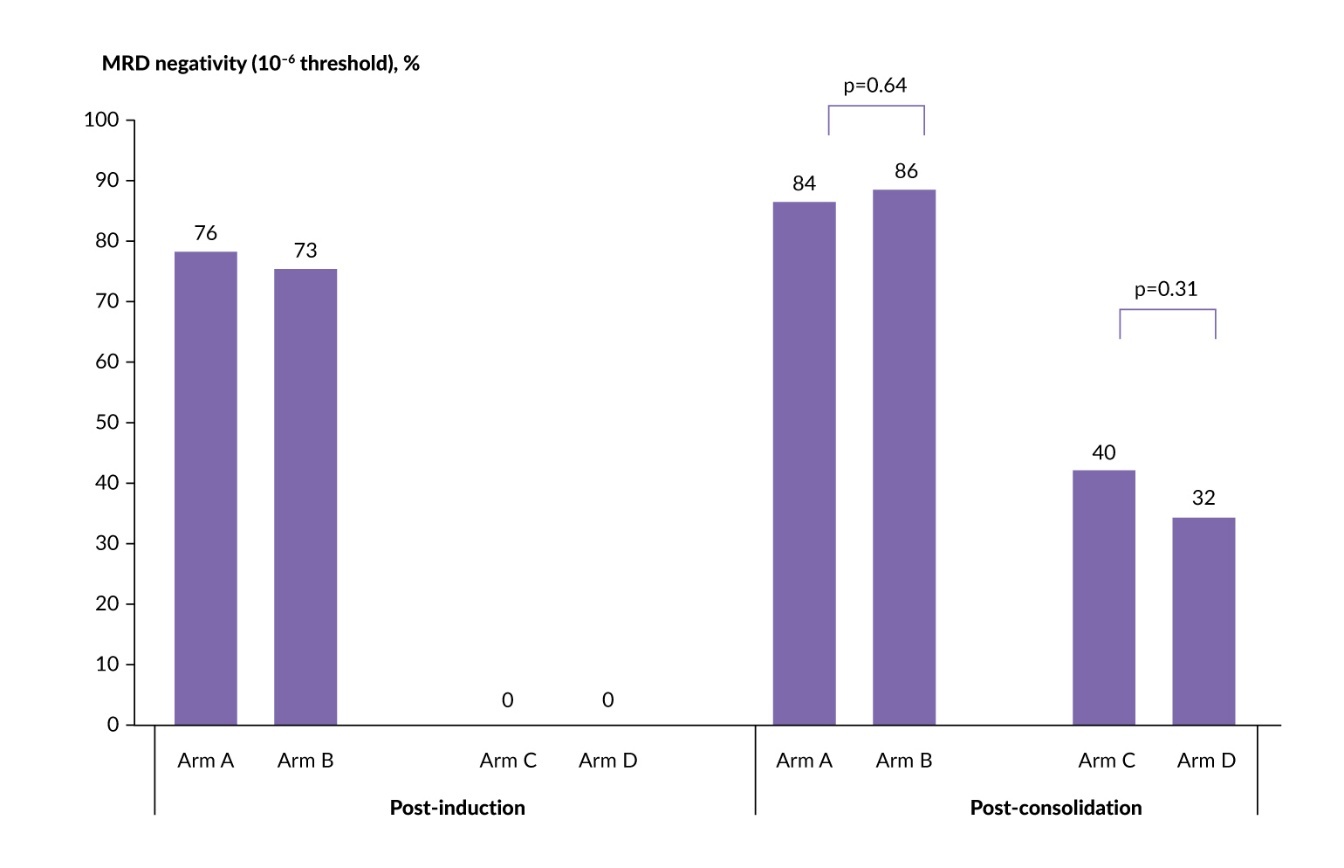
MIDAS aimed to evaluate risk-adapted MRD-driven strategies following Isa-KRd induction and the potential benefit of high-dose melphalan–ASCT.35 TE patients with NDMM received six cycles of Isa-KRd, after which MRD status was assessed (10–5 by NGS). Standard-risk patients (MRD <10–5) were randomized 1:1 to six additional cycles of Isa-KRd (arm A) or ASCT followed by two cycles of Isa-KRd (arm B). High-risk patients (MRD ≥10–5) were randomized 1:1 to single ASCT followed by two cycles of Isa-KRd (arm C) or tandem ASCT (arm D). Maintenance therapy included three years of lenalidomide in the ASCT and Isa-KRd groups and isatuximab plus iberdomide in the tandem and single ASCT groups. The primary endpoint was post-consolidation MRD negativity at 10–6 sensitivity.
Of 791 enrolled patients, 63% attained MRD negativity at 10–5 following Isa-KRd induction.35 Remarkably, high-dose melphalan–ASCT showed efficacy comparable to continued Isa-KRd in the standard-risk cohort, with post-consolidation MRD negativity rates at 10–5 of 86% versus 84% (adjusted relative risk [RR]: 1.02 [95% CI: 0.95–1.10]; p=0.64). In the high-risk cohort, tandem transplantation did not provide additional benefit over single ASCT (MRD negativity, 32% vs 40%; adjusted RR: 0.82 [95% CI: 0.58–1.15]; p=0.31) (Figure 2). Notably, 15% of patients assigned to the tandem ASCT arm did not proceed to the second transplant. The safety profile during consolidation was consistent with prior studies, with higher rates of mucosal inflammation and stomatitis in the ASCT arms.
_negativity_in_the_midas_trial.jpeg)
The MIDAS results are practice-changing and help address critical questions regarding the necessity of high-dose melphalan–ASCT for patients who achieve MRD negativity after induction,70 demonstrating that MRD status has the potential to move from a purely prognostic marker to a functional, real-time clinical decision-making tool. For patients who achieve MRD negativity after modern quadruplet induction, high-dose melphalan–ASCT may be safely omitted. At the same time, subgroup analyses from MIDAS did show improved MRD negativity with ASCT in certain high-risk cytogenetic subgroups, including t(4;14),35 supporting a selective, risk-adapted approach.
Bispecific antibodies
Despite the success of anti-CD38 quadruplets, approximately half of NDMM patients still do not achieve MRD negativity. Moreover, patients with high-risk cytogenetics, extramedullary disease or frailty have suboptimal outcomes with existing regimens, motivating the earlier integration of next-generation immunotherapies. B-cell maturation antigen (BCMA) × CD3-directed bispecific antibodies teclistamab, linvoseltamab and elranatamab have all demonstrated deep and durable responses in RRMM and are now being evaluated in NDMM.
Teclistamab
Teclistamab, a first-in-class bispecific BCMA × CD3-targeting antibody, was investigated in the MajesTEC program across different treatment settings. In RRMM, deep and durable responses were demonstrated in the MajesTEC-1, MajesTEC-2 and MajesTEC-3 trials, including heavily pretreated patients.71–73 MajesTEC-437 and MajesTEC-536 evaluated teclistamab in NDMM, focusing on maintenance and induction therapies, respectively, whereas IFM2021-0139 assessed teclistamab in elderly TIE patients.
MajesTEC-5: Teclistamab-based induction in TE patients
The phase II MajesTEC-5 study evaluated teclistamab- and talquetamab-based regimens in TE NDMM. Recent analysis summarized outcomes from three cohorts of patients (n=49) receiving teclistamab induction.36 Patients received D-Rd in combination with either teclistamab 1.5 mg/kg weekly (arm A) or teclistamab 3.0 mg/kg every four weeks (Q4W) (arm A1), or D-RV plus teclistamab 3.0 mg/kg Q4W (arm B). The primary endpoint was safety and the secondary endpoints were response rates and MRD negativity at 10−5. Remarkably, 100% of evaluable patients achieved MRD negativity after cycle 3, with an overall response rate (ORR) of 89.5–100% (≥CR, 52.6–100%) across the three arms. Safety profile was manageable and consistent with those known for the individual components. These data support the feasibility of combining teclistamab with established quadruplets to further deepen responses in NDMM.
MajesTEC-4: Teclistamab-based maintenance following ASCT
Previous studies suggested that adding bispecific antibodies to IMiD maintenance may improve responses following ASCT due to the synergistic cytotoxic and immunomodulatory effects of each agent.74,75 The phase III MajesTEC-4 trial evaluated fixed-duration teclistamab maintenance with or without lenalidomide versus lenalidomide alone in NDMM patients (n=94) who achieved a partial response or better (≥PR) after a triplet or quadruplet induction regimen, followed by ASCT and consolidation.38 The primary endpoints were PFS and MRD-negative CR at 12 months. In the safety run-in phase, all cohorts achieved an unprecedented 100% MRD negativity during maintenance, with ≥CR rates of 90.6–100% at a median follow-up of 21.1 months and median PFS not reached. Grade 3–4 TEAEs were reported in 56.7–100% of patients across cohorts; however, treatment discontinuation rate due to TEAEs was low (5.3%) and grade 3–4 cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS) were not observed. These results suggest that adding teclistamab to maintenance achieves high MRD negativity rates, potentially allowing for fixed-duration therapy rather than long-term lenalidomide.
IFM2021-01: Teclistamab plus daratumumab in elderly TIE patients
In the phase II IFM2021-01 TecLille study evaluating teclistamab in combination with daratumumab or lenalidomide in elderly TIE patients with NDMM, all 37 patients treated with teclistamab plus daratumumab achieved 100% ≥VGPR as best response at a median follow-up of 10.3 months, with a ≥VGPR rate of 79%.39 MRD negativity (10–6) was achieved in all 27 evaluable patients and in 73% of the intention-to-treat (ITT) population. Both PFS and OS rates were 100%, with no grade ≥3 CRS and grade ≥3 infections, supporting chemotherapy-free teclistamab-based doublet regimens in frail or TIE patients.
Linvoseltamab
LINKER-MM4: A simplified monotherapy with linvoseltamab
The phase I/II LINKER-MM4 study evaluated linvoseltamab as a simplified first-line monotherapy in both TE and TIE NDMM patients (n=45).40 Efficacy was encouraging, particularly at the 200 mg dose, with an ORR of 86% and ≥CR rates of 43%. All MRD-evaluable patients receiving the 200 mg dose were MRD negative at 10–5; across all doses, 95% of patients achieved MRD negativity at 10–5. Linvoseltamab demonstrated a manageable safety profile across all doses, with no dose-limiting toxicities or grade 5 TEAEs.
IMMUNOPLANTTM: Fixed-duration immunoconsolidation with linvoseltamab
The phase II IMMUNOPLANT study used a short, fixed-duration (4–6 cycles) linvoseltamab immunoconsolidation in patients who achieved ≥VGPR but remained MRD positive after standard triplet or quadruplet therapy.41 The primary endpoint was MRD conversion rate at 10−6. Among 18 evaluable patients, 100% converted to MRD negativity (10−6); notably, 17 of 19 patients achieved MRD negativity after only four cycles of linvoseltamab and all remained MRD negative at six months. Linvoseltamab was generally well tolerated, with low rates of grade 3 TRAEs and no CRS or ICANS reported with step-up dosing and prophylactic tocilizumab.
Elranatamab
The phase III MagnetisMM-6 study evaluated elranatamab in combination with daratumumab and lenalidomide versus D-Rd in TIE NDMM patients.42 Initial results from part 1 dose level G showed a confirmed ORR of 97.3% and a ≥VGPR rate of 94.6% at a median follow-up of 7.9 months, with a manageable safety profile, supporting further evaluation of elranatamab-based dexamethasone-sparing combinations in this setting.
Antibody-drug conjugates
Belantamab mafodotin
DREAMM-9: Belamaf plus RVd in TIE patients
Belantamab mafodotin (belamaf) is a first-in-class ADC targeting BCMA with a potent maleimidocaproyl monomethyl auristatin F (mcMMAF) cytotoxic payload.76 Belamaf-based combinations have demonstrated significantly prolonged PFS compared with SoC in RRMM.77,78 The phase I DREAMM-9 trial evaluated belamaf plus RVd combination in TIE patients with NDMM (n=108) across eight dosing cohorts.43 The primary endpoint was safety. The addition of belamaf to RVd resulted in very high ORRs (71–100%), with 62–92% achieving ≥CR. Higher starting doses and shorter dosing intervals were associated with higher MRD negativity rates that continued to improve during the maintenance phase. Most patients experienced keratopathy and visual acuity (KVA) AEs (grade 3–4, 55%); however, ocular toxicity was generally manageable through dose modifications, with low discontinuation rates (5%). The phase III DREAMM-10 study evaluating belamaf plus Rd versus D-Rd in TIE patients with NDMM is currently recruiting.79
Management of belamaf-associated ocular events
Ocular events are a frequent and potentially dose-limiting toxicity of belamaf,80 with practical guidance on its management underscoring the importance of a multidisciplinary approach.81,82 Ophthalmologic evaluation is recommended before each of the first four treatment cycles, after which the Vision-Related Anamnestic (VRA) questionnaire may be used to monitor symptoms and guide decisions. Dose adjustments are based on event severity per the KVA scale, with dose reduction to 1.9 mg/kg or extension of dosing intervals to every 8–12 weeks.
The utility of the VRA tool and modified dosing strategies were explored in recent trials. In the phase I/II BelaRd study evaluating belamaf plus Rd in TIE, intermediate-fit or frail patients, safety was manageable at the recommended phase II dose (1.9 mg/kg) and the rates of grade ≥3 OEs were comparable across groups.44 Similarly, the phase I/II BelaDRd trial evaluated belamaf (1.9 or 1.4 mg/kg) administered every 8 or 12 weeks in combination with D-Rd.45 The regimen demonstrated manageable toxicity profile with high efficacy: 87.5% of patients achieved a PR and a 1-year PFS rate was 90.7%.
CELMoDs
Next-generation CELMoDs bind to cereblon, a ubiquitin ligase (E3) substrate receptor, with higher affinity than classic IMiDs, resulting in potent anti-myeloma activity.83–85 Preclinical studies demonstrated synergism between iberdomide and standard MM therapies.86,87 In the phase I/II CC-220-MM-001 trial, CELMoD iberdomide was investigated as monotherapy and in combination with Vd in NDMM or RRMM .46,48,49 Among 18 patients for whom ASCT was not planned or recommended, iberdomide plus Vd demonstrated an ORR of 88.9% in the dose expansion phase. In the updated analysis, responses deepened over time, with two-thirds of patients achieving a CR or sCR; MRD negativity at 10–5 was reported in 44.4% of patients, all of whom achieved ≥CR.47 Grade 3–4 TEAEs occurred in 82% of patients, primarily hematologic (59%) and infections (47%), with infrequent discontinuations due to toxicity. Additional evidence was provided by the phase II GEM-IBERDARAX trial in TIE patients (n=73), most of whom were frail or ultrafrail. The ORR with the triplet regimen of iberdomide, daratumumab and dexamethasone was 93.1% (sCR, 30.1%) and the safety profile was acceptable and manageable.50 The ongoing phase III EXCALIBER study investigates iberdomide versus lenalidomide as maintenance after ASCT.88 The primary endpoint is PFS, with key secondary endpoints including OS, response rates, MRD negativity, safety and health-related QoL. This study will be critical to determine whether next-generation CELMoDs can improve the depth and durability of response compared with lenalidomide, which remains the current SoC for maintenance in NDMM.
Genetic, molecular and other biological determinants of treatment outcomes
Beyond novel agents, understanding genetic determinants of treatment response is essential for personalized therapy. Two recent analyses provide biological insights that may guide selective, risk-adapted treatment approaches in NDMM.
The impact of Duffy genotype: Exploratory analysis of DETERMINATION
A recent analysis provided a biologically driven explanation for previously observed differences in PFS by race in the phase III DETERMINATION trial, which randomized TE patients with NDMM (n=722) to RVd alone (n=357) or RVd plus upfront ASCT (n=365), followed by continuous lenalidomide maintenance.51 At a median follow-up of 76 months, RVd plus ASCT demonstrated a PFS benefit versus RVd alone (HR: 1.53 [95% CI: 1.23–1.91]); however, no significant OS benefit was observed. Subgroup analyses showed that the PFS benefit with RVd plus ASCT was maintained in White patients, whereas PFS was similar between the treatment arms in African American patients.
The investigators explored potential genetic contributors to outcome heterogeneity, focusing on the erythrocyte Duffy null phenotype which results from a single nucleotide polymorphism in the DARC/ACKR1 chemokine receptor gene.89 This phenotype is highly prevalent in individuals of African ancestry, is associated with lower absolute neutrophil count, plays a key role in cytokine homeostasis and is believed to confer an evolutionary advantage by reducing susceptibility to Plasmodium vivax infection. Among 493 randomized patients with genomic data, 12% were Duffy null, the vast majority of whom were African American(~90%).51 In a pooled analysis, PFS was similar between Duffy null and non-null patients; however, marked heterogeneity emerged when outcomes were examined by treatment arm. In Duffy non-null patients, PFS findings mirrored the ITT results, with a clear benefit favoring RVd plus ASCT (HR: 1.76 [95% CI: 1.33–2.34]; p=0.005 favoring ASCT). In contrast, Duffy-null patients showed numerically longer PFS with RVd alone (HR: 0.64 [95% CI: 0.27–1.50]; interaction p=0.005). Univariate and multivariable analyses further showed that Duffy status was a stronger prognostic factor for PFS in the RVd-alone arm than race itself.
These data are consistent with MIDAS findings,35 suggesting that the clinical value of high-dose melphalan–ASCT is confined to selected patient subgroups. The pronounced PFS benefit in the absence of OS benefit in DETERMINATION is most consistent with competing risks and treatment effect heterogeneity, with disease control achieved in a proportion of patients, while others experience adverse consequences, limited benefit, or both, such that any survival advantage is neutralized. These considerations are particularly relevant for melphalan, which is genotoxic to both malignant plasma cells and normal hematopoietic stem and progenitor cells. Therapy-related secondary leukemia and myelodysplastic syndromes represent a clinically important competing risk of alkylator-induced DNA damage.90,91 Relapse after high-dose melphalan is characterized by increased genomic complexity and treatment resistance, which may indicate mutational accumulation and clonal selection under alkylator pressure. Other datasets demonstrate inferior outcomes of CAR T-cell therapy in patients previously exposed to high doses of alkylators,92 which is compatible with adverse tumor biology at relapse, including increased mutational burden and resistant clonal architecture. Taken together, these observations support a selective, risk-adapted approach to high-dose melphalan and transplantation in NDMM.
MRD trajectory in NDMM with t(11;14) translocation
MM harboring t(11;14)(q13;q32) translocation represents a biologically distinct entity characterized by more oligo-secretory phenotype, frequent CD20 expression and greater dependence on BCL2 signaling. Clinically, it is associated with slower response to therapy, raising the question of whether the depth and dynamics of MRD response to modern quadruplet regimens in these patients mirror those of t(11;14)-negative disease. A retrospective analysis characterized the trajectory of MRD and treatment outcomes according to t(11;14) status in NDMM patients who received quadruplet therapy with MRD-adapted consolidation/maintenance and treatment duration.52 The cohort comprised 302 patients treated with D-KRd in the MASTER trial (n=113), D-RVd in the MILESTONE trial (n=11) and D-RVd within a SoC database (n=177). The study showed that t(11;14)-positive patients (16% of cohort) had significantly slower MRD clearance compared with t(11;14)-negative patients: MRD negativity rates at 10–5 were 9% versus 31% after induction, 36% versus 59% after ASCT and 53% versus 75% at any time during follow-up. Despite slower clearance, rates of sustained MRD negativity beyond 12 months were comparable, and 4-year PFS was superior in t(11;14)-positive patients (90% vs 72%). These data illustrate that slower response does not necessarily imply worse outcome, reinforcing the need for biologically informed interpretation of molecular MRD kinetics in various patient subgroups.
Conclusions
The MM treatment landscape has rapidly advanced in recent years. Quadruplet regimens incorporating anti-CD38 mAbs have significantly improved clinical outcomes compared with standard triplet therapies in patients with NDMM, including fit elderly individuals and those who are ineligible for or have deferred ASCT, as demonstrated in phase III trials. Early-phase trials suggest high potential of incorporating novel immunotherapy approaches, including BCMA-directed bispecifics, ADCs and CELMoDs, early in the treatment strategy. While these efficacy signals are encouraging, further studies are necessary to unequivocally establish their efficacy. Practice-changing recommendations should await confirmation from larger ongoing or planned trials. Furthermore, safety profiles of novel agents require careful consideration. For instance, grade 3–4 infections are observed in approximately 30% of bispecific antibody recipients, often requiring prophylaxis, including immunoglobulin replacement, with neutropenia being the most common grade ≥3 haematological toxicity (pooled incidence up to approximately 50%).36,38,93 These data underscore the need for comprehensive toxicity management and supportive care when integrating bispecifics into treatment protocols, especially for older and frail patients.
Although CAR T-cell approaches were not within the primary scope of this review, their potential role in NDMM should also be noted. Ongoing randomized studies, including CARTITUDE-594 in patients for whom ASCT is not planned as initial therapy and CARTITUDE-695 in TE patients, are evaluating whether ciltacabtagene autoleucel can improve outcomes compared with current frontline strategies.
MRD is emerging both as a prognostic marker and potential decision-making tool in NDMM. However, MRD has limitations as a surrogate endpoint, particularly in the absence of mature OS data. The optimal sensitivity threshold, timing of assessment and duration of MRD negativity required to predict long-term benefit are not yet standardized. Therefore, MRD status should be considered together with established clinical features and cytogenetic risk stratification markers.
The use of high-dose melphalan and ASCT remains a key discussion point in NDMM management. While data from the MIDAS trial supported de-escalation or omission of ASCT in MRD-negative patients, the DETERMINATION trial did demonstrate a significant PFS benefit with upfront ASCT, although no OS advantage suggesting possible competing risk. Taken together, these divergent results likely reflect differences in induction intensity, patient populations and backbone regimens, as well as the importance of avoiding a “one size fits all” philosophy to treatment selection. Rather than favoring a universal approach, the current evidence supports a selective, risk-adapted strategy: ASCT may be omitted and/or deferred in patients who achieve deep MRD negativity after highly effective quadruplet induction. Conversely, it may still provide benefit in those with persistent MRD positivity and/or certain high-risk cytogenetic or clinical features. Longer follow-up for OS in current studies and prospective validation across quadruplet induction regimens with incorporation of novel immunotherapies (including bispecifics, ADCs and CAR T-cell therapy) are underway and will be needed to establish the optimal approach in different patient populations.
Ongoing clinical research aims to further optimize patient outcomes by refining treatment algorithms and guidance strategies, improving risk stratification to balance efficacy against tolerability, and incorporating novel agents into established regimens, with the goal of successful translation to real world practice.96
Acknowledgements
The authors gratefully acknowledge the editorial support and assistance of healthbook in the preparation of this manuscript.
Conflict of interest
Yuxin Liu has participated in the advisory board for Opna Bio. Taya Jamal Salman, Tarek H. Mouhieddine, Poy Theprungsirikul, Isabella Ruiz, Brendan Fink, Keren Bobilev and Jacob Laubach have no conflicts to disclose. Clifton Mo acted on the advisory boards for AbbVie, BMS, GSK, Janssen, Karyopharm, Sanofi and Takeda, and as a consultant for AbbVie, Janssen, Karyopharm and Sanofi. Shonali Midha reported Abbvie stock ownership and honoraria for consultancy from Janssen and Pfizer. Omar Nadeem reported advisory board participation for JNJ, BMS, Sanofi, GPCR therapeutics, Kite and AstraZeneca, and research funding from BMS and JNJ. Paul G. Richardson received honoraria for consultancy from Celgene/BMS, GSK, Karyopharm, Oncopeptides, Regeneron and Sanofi, and research grants from Oncopeptides. These funding entities did not play a role in the development of the manuscript and did not influence its content in any way.
Funding
The authors have declared that no financial support was received from any organization for the submitted work.
Author contributions
The authors have created and approved the final manuscript.
AI use
The authors declared that no generative artificial intelligence (AI) or AI-assisted technologies were used in the preparation or writing of this manuscript. All content was produced entirely by the authors who take full responsibility for the accuracy, integrity and originality of the work.