Introduction
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) characterized by the accumulation of abnormal promyelocytes and balanced translocation t(15;17)(q24;q21), which results in the formation of the PML::RARA oncogene. A small proportion of patients harbor variant translocations that lead to fusion of RARA with one of several alternative partner genes.1,2 The most frequently reported variant is the t(11;17)(q23;q21), resulting in fusion of the zinc finger gene ZBTB16 (formerly PLZF) with the RARA locus (ZBTB16::RARA).3
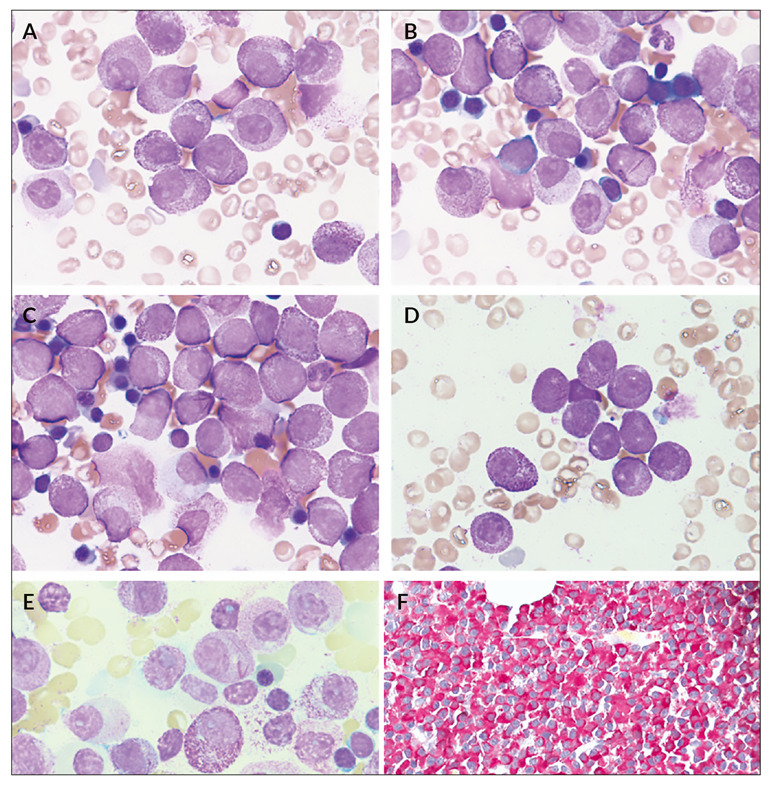
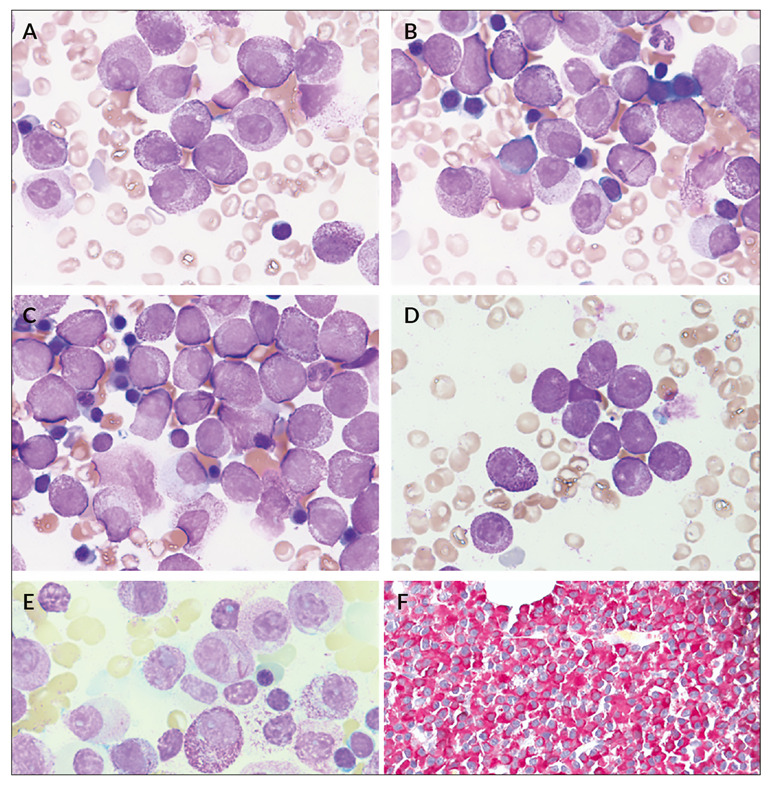
Typical APL is morphologically characterized by abnormal promyelocytes with irregular and highly variable nuclei, which are often kidney-shaped or bilobed in variant APL, and densely packed cytoplasm with large granules and single or bundled (“faggots”) Auer rods. In contrast, cases with the variant RARA translocation t(11;17)(q23.2;q21.2) resulting in ZBTB16::RARA show distinct cytomorphological features, including a predominance of cells with regular nuclei, abundant granulations, frequent absence of Auer rods and an increased number of pelgeroid neutrophils.2,4 However, a review of the literature indicates that the absence of Auer rods or faggot cells, previously described as a characteristic morphologic feature of t(11;17)(q23;q21) APL,2,4,5 cannot be confirmed in all published cases. Granularity is also highly variable and therefore not a reliable distinguishing feature from classic or variant APL with t(15;17)/PML::RARA.
When the hypercoagulable status is controlled by supportive therapy, typical APL has a good prognosis and responds well to retinoid-based therapy with all-trans retinoic acid (ATRA) in combination with either anthracyclines or arsenic trioxide (ATO), which yields high rates of complete remission and overall survival.6,7 Therefore, it is essential to recognize variants of RARA translocations, as the fusion partner considerably impacts disease biology, particularly with regard to ATRA sensitivity.
APL can be divided into two disease subtypes: an ATRA-responsive subtype, which includes APL with RARA fused to PML, NPM1, NuMA, and possibly BCOR, PRKAR1A or FIP1L1; and ATRA- and ATO-resistant subtype characterized by the presence of the ZBTB16::RARA and STAT5B::RARA fusions.1,5,8 Although ATRA induces degradation of the ZBTB16–RARA fusion protein at the molecular level, oncoprotein loss is insufficient to induce differentiation or clinical response.9 Resistance to ATRA has been attributed to the reciprocal RARA–ZBTB16 proteins which upregulates CRABPI, a retinoic acid-binding protein involved in retinoid catabolism.10
Most variant RARA rearrangements, including RARA::ZBTB16, are resistant to anthracyclines and ATO, and combinations of ATRA with anthracycline have generally not been successful.11 Prolonged exposure to higher-dose ATRA (60 mg/m2 instead of 45 mg/m2) has shown limited differentiation effects, suggesting that response to ATRA was not completely lost.11 Some clinical benefit has been observed when combining ATRA and other agents, such as granulocyte colony-stimulating factor (G-CSF) or histone deacetylase inhibitors.11–14 Nevertheless, most patients relapse within one year and subsequently show refractoriness to further therapy. Standardized treatment recommendations are lacking due to the rarity of these variants.
To date, high-dose chemotherapy consisting of busulfan and cyclophosphamide followed by autologous stem cell transplantation has not been described as a consolidation strategy in this setting. Here, we present a case of a patient with ZBTB16::RARA-positive APL who achieved a durable molecular complete remission lasting more than two years following high-dose chemotherapy with busulfan and cyclophosphamide and subsequent autologous stem cell transplantation.
Clinical presentation
Initial presentation and diagnosis
A 68-year-old patient initially presented with slowly progressive pancytopenia over a six-month period. Peripheral blood counts at diagnosis showed neutrophils 0.42 Gpt/L, hemoglobin 8.7 g/dL and platelets 88 Gpt/L). He was in very good general health without signs of bleeding. The only relevant comorbidity was hereditary hemochromatosis.
Peripheral blood smear revealed 12% hypergranulated promyelocytes, including one containing faggots of Auer rods, contrary to what is reported in the literature.4,15 Bone marrow aspirate showed predominantly hypergranulated promyelocytes with mostly round, non-lobated nuclei (Figures 1A-E). Auer rods and faggots were present. Few neutrophilic granulocytes showed pseudo-Pelger-Huët anomalies and occasional cytoplasmic inclusions.
Molecular testing did not confirm the suspected PML::RARA-transcript. Cytogenetic analysis revealed a reciprocal translocation between the long arm of chromosome 11 and the long arm of chromosome 17 in 10 of 20 metaphases, corresponding to a ZBTB16::RARA rearrangement. The remaining metaphases showed a normal male karyotype. Interphase fluorescence in situ hybridization (FISH) detected the RARA rearrangement in 92 of 100 cells.
Therapeutic interventions and outcomes
Given the known resistance of ZBTB16::RARA APL to ATRA, ATO1,7 and standard-dose cytarabine and daunorubicin chemotherapy, we initiated induction therapy consisting of intermediate-dose cytarabine (1,000 mg/m2 twice daily on days 1–3), mitoxantrone (10 mg/m2 on days 3–5), ATRA (45 mg/m2 on day 1 up to day 60) and G-CSF (5 µg/kg). G-CSF was included based on reports suggesting partial reversal of ATRA resistance.1,13 Dexamethasone (10 mg/m2) was administered prophylactically during the first two weeks to prevent differentiation syndrome, followed by slow dose tapering.
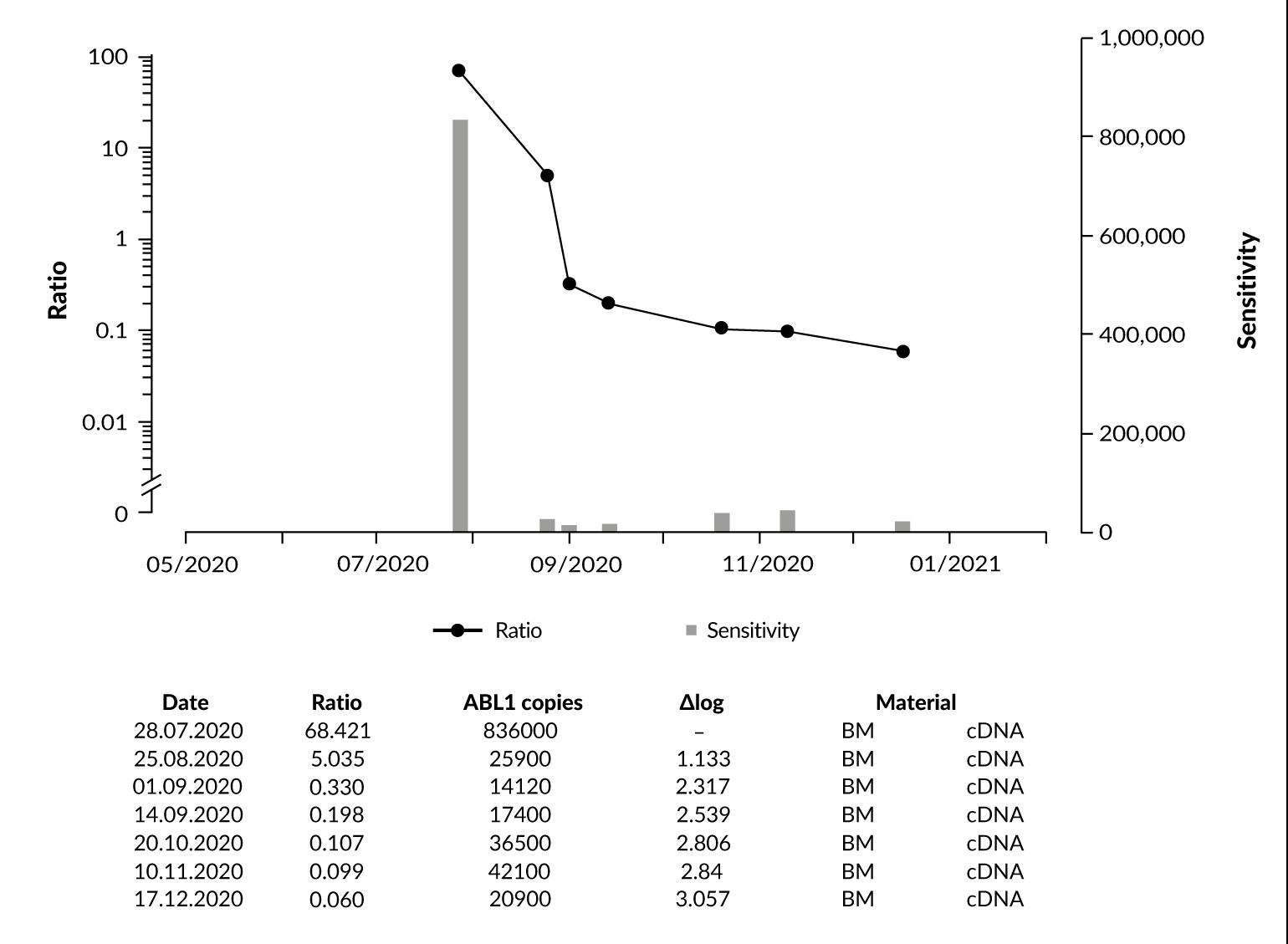
Bone marrow evaluation on day +28 showed a good molecular response, with quantitative PCR for ZBTB16::RARA decreasing from an initial percentage of 68.421% to 5.035% (Figure 2). Following induction therapy, autologous stem cell apheresis from peripheral blood was successfully performed. The apheresis product was negative for ZBTB16::RARA.
_monitoring_of_*zbtb16__rara*_trans.png)
Relevant side effects included a generalized maculopapular rash, which required continuous treatment with prednisolone and antihistaminic drugs, as well as severe bone pain predominately affecting the hips and lumbar spine. Infectious or extramedullary disease was excluded by computed tomography (CT) and magnetic resonance tomography (MRT) imaging and laboratory evaluation. These symptoms were considered potentially related to the combination of twice-daily G-CSF and concomitant ATRA. After completion of G-CSF and ATRA therapy, the rash resolved, while bone pain persisted for several weeks at reduced intensity.
A bone marrow aspirate performed before the first consolidation therapy with intermediate-dose cytarabine (1 g/m2 twice daily on days 1,3 and 5) in combination with ATRA (45 mg/m2 daily) showed further molecular response, with the ZBTB16::RARA ratio declining to 0.330% (Figure 2). During this phase, the patient again developed generalized rash, fever and pain. G-CSF was discontinued and ATRA was continued until day +21.
Further evaluation suggested that a macrophage-activating syndrome (MAS) may have contributed to recurrent fever, pain and rash. Sweet syndrome and relevant infectious complications were repeatedly excluded.
The second consolidation therapy consisted of high-dose chemotherapy with busulfan (4 × 0.8 mg/kg/day on days -7 to -4) and cyclophosphamide (60 mg/kg on days -3 to -2), followed by autologous stem cell transplantation (3.88 × 106 CD34+ cells/kg) on day 0. Before transplantation, the absence of leukemic contamination in the stem cell apheresis product was confirmed by PCR.
The therapy was well tolerated. Serial assessments of minimal residual disease (MRD) showed complete molecular remission. Figure 2 illustrates further progressive decline of ZBTB16::RARA expression following consolidation therapy and high-dose chemotherapy with autologous stem cell transplantation. The patient remains in sustained complete remission for nearly two years after transplantation.
Discussion and conclusions
A genetic variant of APL which does not involve the PML::RARA gene fusion is very rare and constitutes <2% of all APL cases. Currently, 17 distinct gene fusion partners of RARA have been described that resulted in the development of an APL or APL-like disease.1,16 Within the group of genetic APL variants, APL caused by the ZBTB16::RARA gene fusion is the most prevalent, accounting for approximately 1% of all APL cases.3,17 This genetic APL variant is resistant to treatment with ATRA. In ZBTB16::RARA APL, the function of the lineage-determining transcription factor CCAAT/enhancer-binding protein alpha (CEBPa) appears to be strongly impaired.18 Guidez et al. demonstrated that the reciprocal translocation product RARA-ZBTB16 leads to increased expression of cellular retinoic acid-binding protein I (CRABPI), a receptor involved in the catabolism of retinoids,10 which might explain the strongly reduced susceptibility to ATRA and why the application of higher ATRA doses in these cases may at least partially overcome resistance to therapy.
Given the scarcity of prospective data on ZBTB16::RARA APL, the reported management has largely relied on AML-like induction and intensified consolidation rather than differentiation-based regimens. A large series of X-RARA cases indicate that the outcomes for ZBTB16::RARA are substantially inferior to those for PML::RARA APL treated with ATRA/ATO, supporting the early consideration of treatment intensification and transplant-based strategies in selected patients.19,20 In this context, achieving a deep molecular response prior to consolidation is clinically relevant because transplant outcomes, particularly with autologous approaches, appear best when performed in molecular remission,21 providing a rationale for the durable remission observed in our patient.
If allogeneic stem cell transplantation is not an option, this case report suggests high-dose chemotherapy with autologous stem cell transplantation as an effective consolidation therapy for patients with ZBTB16::RARA variant APL. The acceptable safety profile and durable molecular remission observed in our patient suggest that this therapeutic approach may help overcome treatment resistance in this rare APL variant. Future studies incorporating molecular profiling of additional mutations may further refine risk stratification and enable individualized therapeutic management for patients with ZBTB16::RARA variant APL. In 2025, the patient remained alive and in remission.
Ethics approval and consent to participate
Written consent for the further use of patient data was obtained.
Consent for publication
Consent for publication was obtained.
Availability of data and materials
All patient data that support this case report are included in anonymized form in the published article.
Conflict of interest
The authors declared that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors declared that no financial support was received from any organization for the submitted work.
Author contributions
All authors contributed substantially to this work. The specific contributions are as follows: SP and MS wrote the manuscript. GO, TH and KSK contributed to the diagnostic course and drafting the manuscript and commented on the manuscript. All authors approved the final manuscript.