




Introduction
Prostate-specific membrane antigen (PSMA), first described in 1987 as a prostate tumor-associated membrane protein, has since become a validated target for molecular imaging in prostate cancer.1 The initially and most widely adopted PSMA-targeting radiotracer in clinical practice was 68Ga-PSMA-11, extensively studied in prospective multicenter trials conducted at the University of California, Los Angeles (UCLA) and the University of California, San Francisco (UCSF).2–4 These investigations demonstrated its clinical utility in both initial staging and recurrence detection. In the preoperative setting, 68Ga-PSMA-11 achieved a sensitivity of 40% and specificity of 95% for detecting pelvic lymph node metastases, with a positive predictive value (PPV) of 75% and negative predictive value (NPV) of 87%.5 In patients with biochemical recurrence, detection rates correlated with prostate-specific antigen (PSA) levels, ranging from 38% in patients with PSA <0.5 ng/mL to 97% in those with PSA ≥5.0 ng/mL.6 These findings led to U.S. Food and Drug Administration (FDA) approval in 2020. However, limitations such as the short half-life, declining generator yields and manufacturing challenges restricted its broader use.7
To overcome these logistic barriers, 18F-labeled PSMA ligands were developed, enabling scalable cyclotron production and wider availability. Among them, 18F-DCFPyL, a second-generation radiotracer, demonstrated normal tissue biodistribution patterns similar to 68Ga-PSMA-11, with comparable uptake in the kidneys and bladder, and low background activity.7 While 68Ga-PSMA-11 exhibited significantly higher uptake in the kidneys, spleen and major salivary glands, 18F-DCFPyL showed slightly greater liver uptake (Figure 1). These similarities established their interchangeability in clinical practice.7 Further comparative studies reinforced these findings. A retrospective study of 80 patients receiving 177Lu-PSMA-617 therapy found no significant differences in biodistribution or lesion uptake between the two tracers, although 18F-DCFPyL demonstrated higher uptake in visceral metastases.8 Based on findings from the phase III OSPREY and CONDOR trials,9,10 18F-DCFPyL is currently approved by Swissmedic, the European Medicines Agency (EMA) and FDA for positron emission tomography (PET) imaging of PSMA-positive lesions in men with prostate cancer.11–13
_radioligan.jpg)
Another major advance came with 18F-radiohybrid (rh)PSMA-7.3 (18F-flotufolastat), which received FDA approval following the phase III LIGHTHOUSE and SPOTLIGHT trials.15–17 In the LIGHTHOUSE study, which enrolled newly diagnosed patients planned for prostatectomy, 18F-rhPSMA-7.3 demonstrated high specificity (93−97% across all readers) and a favorable safety profile, although it did not meet the coprimary endpoint of patient-level sensitivity.16 In the SPOTLIGHT study, the majority-read detection rate was 83% for recurrent prostate cancer localization, but the trial did not meet the coprimary endpoint of combined region-level PPV, defined as the accuracy of lesion-level detection confirmed by histopathology, imaging or clinical follow-up across multiple anatomical regions.17 A predefined exploratory analysis, however, confirmed high detection accuracy in patients with negative baseline conventional imaging, with true-positive lesions identified in 64% of patients.18 Subgroup analyses showed detection in the prostate bed, pelvic lymph nodes and distant sites. While these data suggest that 18F-rhPSMA-7.3 outperforms conventional imaging and older PET radiotracers such as choline or 18F-fluciclovine, no direct comparison with 68Ga-PSMA-11 and 18F-DCFPyL has been reported.19 Table 1 summarizes the main findings of the pivotal trials on the three FDA-approved imaging radiotracers in the biochemically recurrent prostate cancer setting. A comparative analysis reveals a PPV of 92% for 68Ga-PSMA-11 in the UCLA/UCSF trial, a correct localization rate of 85−87% for 18F-DCFPyL in the CONDOR trial and a verified detection rate of 57% and a combined region-level PPV of 60% for 18F-rhPSMA-7.3 in the SPOTLIGHT study. Figure 1 presents normal body distribution of 18F-rhPSMA-7.3.
The 18F-PSMA-1007 radiotracer represents another promising second-generation agent, characterized by low urinary excretion. 18F-PSMA-1007 demonstrated high detection rates both in primary and biochemically recurrent prostate cancer after radical prostatectomy,20,21 with superiority over multiparametric magnetic resonance imaging (MRI) for locoregional staging in intermediate and high-risk patients.22 In a phase III study comparing 18F-PSMA-1007 with 18F-fluorocholine for detecting biochemical recurrence, 18F-PSMA-1007 achieved significantly higher correct detection rates (0.82 vs 0.65 when undetermined findings were considered malignant; 0.77 vs 0.57 when considered benign; both p<0.0001).23
Unlike most urea-based radiotracers, 18F-CTT1057 is built on a phosphoramidate scaffold and binds to PSMA irreversibly. This design resulted in biodistribution patterns comparable to urea-based radiotracers, with reduced exposure to the kidneys and salivary glands and improved sensitivity for metastatic lesions.24 In the phase II/III GuideView trial in newly diagnosed high-risk prostate cancer, 18F-CTT1057 achieved patient-level sensitivity of 86.8–90.0%; and region-level specificity of 97.1% for detection of PSMA-positive lesions, with all confidence intervals surpassing prespecified success thresholds.25,26 Similarly, in the phase III GuidePath study of patients with biochemically recurrent prostate cancer, 18FCTT1057 demonstrated region-level correct localization rate of 65.2–75.0% and patient-level PPV of 64.6–76.5%.27 These findings confirmed the diagnostic efficacy of 18FCTT1057, with high reproducibility and reliability.
Selecting the optimal PSMA radiotracer for prostate cancer imaging
While PSMA PET imaging has clearly demonstrated superiority over conventional CT and bone scans in detecting prostate cancer, there remains a lack of comparative studies evaluating the performance of different PSMA ligands such as 68Ga-PSMA-11 and 18F-DCFPyL. Although newer PSMA PET radiotracers may offer improved sensitivity or logistical advantages, it is still unclear whether they outperform these established agents. As the field advances, it becomes increasingly evident that the era of using CT alone and bone scans as the comparator or gold standard in clinical trials should come to an end, paving the way for more meaningful head-to-head comparisons between PSMA PET radiotracers.
One of the few prospective comparison studies examined 68Ga-PSMA-11 versus 18F-PSMA-1007.28 In this study, 50 men underwent imaging with both radiotracers less than four weeks apart. Results showed that 18F-PSMA-1007 uptake was significantly higher than 68Ga-PSMA-11 uptake in local recurrences, nodal and distant metastases and most physiological sites. However, urinary bladder uptake was significantly lower with 18F-PSMA-1007, which reduces potential urinary-related artifacts. Despite these advantages, 18F-PSMA-1007 imaging led to more frequent upstaging and more equivocal results compared with 68Ga-PSMA-11.
Further supporting these findings, a retrospective matched-pair study assessed non-tumor-related uptake and the detection efficacy of 68Ga-PSMA-11 PET/CT versus 18F-PSMA-1007 PET/CT in patients with recurrent prostate cancer after radical prostatectomy.29 The study, which included 102 patients with 204 matched scans, demonstrated that 18F-PSMA-1007 PET detected almost five times more benign lesions with increased PSMA-ligand uptake compared with 68Ga-PSMA-11 PET (245 vs 52); the benign lesion were more frequently observed in ganglia (43% vs 29%), unspecific lymph node (31% vs 42%) and bone lesions (24% vs 27%).
A notable concern with 18F-PSMA-1007 is unspecific bone uptake without morphological correlations, which can be mistaken for metastasis, potentially leading to patient over-staging and inappropriate treatment.29,30 Building on these clinical observations, a retrospective multicenter study aimed to assess unspecific bone uptake, defined as focal mild-to-moderate uptake (SUVmax <10.0) without an evident benign or malignant cause. Unspecific bone uptakes were detected in 51.4% of patients (179/348), most commonly in the ribs (57.5%), pelvis (24.8%) and spine (9.7%).31 Their occurrence was not associated with prostate-specific antigen (PSA), Gleason score, tumor size, age or injected dose. Notably, unspecific bone uptake was significantly more frequent in digital PET/CT scans (82%) than in analog PET/CT scans (40.3%) (p=0.0001), while no significant difference was observed for digital PET/MR (51%) (p=0.1599). In 45% of patients, unspecific bone uptake influenced clinical decision-making and was considered relevant for therapeutic management.
A separate study evaluating the biodistribution for 68Ga-PSMA-11 and 18F-PSMA-1007 further highlighted considerable differences in radiotracer uptake patterns.32 Results showed that 68Ga-PSMA-11 is primarily excreted through the urinary system, with limited clearance via the hepatobiliary system. In contrast, 18F-PSMA-1007 demonstrated significantly higher liver and gallbladder uptake and minimal urinary excretion. In particular, SUVmean for the liver, spleen, salivary glands and bones were significantly higher with 18F-PSMA-1007 PET than 68Ga-PSMA-11 PET (p<0.05) (Figure 2). However, no significant difference between the two radiotracers was observed in blood pool uptake (p=0.153). Based on these results, the recommended reference organ for quantification is the liver for 68Ga-PSMA-11 and the spleen for 18F-PSMA-1007.14,33

A systematic review evaluated the performance of 68Ga-PSMA-11 and various 18F-labeled PSMA radiotracers across clinical settings in prostate cancer, with a focus on biodistribution, diagnostic efficacy and interpretative challenges.34 The review included head-to-head comparisons, matched-pair studies and prospective and retrospective studies published between 2016 and 2021, each enrolling more than 20 patients. Although comparative data were limited and study heterogeneity, non-standardized and lack of uniform methodologies posed challenges, some consistent patterns emerged. 18F-PSMA-1007 demonstrated superior detection of local recurrences due to its low urinary bladder activity, making it particularly useful for evaluating the prostate bed and radiotherapy planning. However, it also produced a higher rate of non-specific findings in bones and ganglia, requiring experienced interpretation. In contrast, 68Ga-PSMA-11 and 18F-DCFPyL were more effective for identifying distant metastases, including bone marrow involvement, with 18F-DCFPyL associated with fewer equivocal skeletal findings and higher inter-reader agreement. Detection rates at low PSA levels (<0.5 ng/mL) were slightly higher for 18F-PSMA radiotracers, particularly 18F-DCFPyL. Lesion-level analysis revealed that 18F-PSMA agents detected more local and bone lesions, while 68Ga-PSMA-11 identified more lymph node metastases. Importantly, the physiological liver uptake of 18F-PSMA-1007 may complicate eligibility assessments for radioligand therapy when liver-based thresholds are used, as in the VISION trial. The authors conclude that no single radiotracer is universally superior; rather, radiotracer selection should be guided by clinical context, availability, interpretative expertise and regulatory factors. Despite current limitations in comparative evidence, PSMA PET has transformed prostate cancer imaging and remains a cornerstone in the molecular diagnosis and management of the disease.
Different PET radiotracers in patient selection for radioligand therapy
In prostate cancer imaging and treatment, particularly with radioligand therapy such as 177Lu-PSMA-617, performing PET/CT scans with various radiotracers plays a crucial role in patient selection and outcome prediction. For 177Lu-PSMA-617 therapy, all patients in the VISION trial underwent 68Ga-PSMA-11 PET/CT scanning,35 while both 68Ga-PSMA-11 and 18F-deoxyglucose (FDG) PET/CT scans were performed in the TheraP study.36 This more stringent criteria in TheraP led to a higher screen failure rate (31% vs 13% in VISION); however, a greater proportion of patients achieved a PSA decline of ≥50% compared with those in VISION (66% vs 46%).35,36
Despite its widespread application in other malignancies, FDG PET/CT imaging has not been considered a primary imaging tool for prostate cancer, largely due to the perception of low FDG uptake in this cancer type.37 The development of novel radiotracers, including 18F-sodium fluoride, 18F-fluorocholine, 11C-choline, 18F-fluciclovine and, more recently, PSMA PET/CT has further compounded this understanding, as these agents have improved sensitivity, specificity and accuracy. While FDG PET/CT provides limited value in the initial staging of prostate cancer, its clinical significance increases in cases of biochemical recurrence, particularly in patients with higher-grade disease (grade 4 or 5) and elevated PSA levels.
As shown in the TheraP study, dual-radiotracer imaging with FDG and PSMA PET/CT significantly improves the accuracy of disease site assessment.36,38 The addition of FDG PET/CT imaging allows for the identification of discordant disease (PSMA-negative/FDG-positive), which is crucial given that the greatest therapeutic benefit from 177Lu-PSMA-617 therapy depends on robust PSMA expression across all disease sites. Taken together, FDG PET/CT imaging has proven to be a valuable tool in the management of advanced prostate cancer, particularly PSMA-negative disease and as a prognostic biomarker in the evolving field of theranostics. However, additional costs and higher burden on the patient as well as the very reassuring data that contrast-enhanced (ce)CT and PSMA PET potentially compensate the additional information provided by FDG PET will likely prevent its wide adoption for therapy selection.39
Across multiple phase II and III clinical trials,35,36,40–43 PSMA PET was used not only to confirm PSMA-positive disease but, importantly, to exclude patients with insufficient or heterogeneous radiotracer uptake. While PSMA PET was a prerequisite in all studies, the specific thresholds and exclusion parameters varied considerably, reflecting differences in clinical aims and disease stages. Most trials defined PSMA positivity by visual or semi-quantitative uptake on 68Ga-PSMA-11 (or 18F-DCFPyL) PET scans; however, the level of stringency differed. While studies such as VISION and PSMAfore relied on qualitative assessments, typically requiring uptake greater than or equal to liver in at least one lesion,35,43 some others, like TheraP, ENZA-p, SPLASH and UpFrontPSMA, applied quantitative SUVmax thresholds to ensure high radiotracer avidity across all disease sites.36,40–42
TheraP and UpFrontPSMA uniquely incorporated dual-radiotracer imaging with FDG PET to exclude patients with discordant lesions, a finding suggestive of aggressive or dedifferentiated tumor biology less likely to benefit from PSMA-targeted therapy.36,40 Additionally, UpFrontPSMA required high-volume disease for enrollment, defined as at least four bone metastases, including one outside the axial skeleton, or visceral involvement.40 While most studies used 68Ga-PSMA-11 for imaging, SPLASH allowed either 68Ga-PSMA-11 or 18F-DCFPyL, balancing broader accessibility with the need for consistent and reliable diagnostic performance.42
These variations in PSMA PET-based eligibility criteria across clinical trials reflects how differences in diagnostic stringency can shape trial populations and treatment outcomes. Trials employing rigorous molecular imaging thresholds, such as requiring high SUVs or excluding patients with discordant lesions (e.g., PSMA-negative but FDG-positive), tend to include patients with uniformly high PSMA expression, who are biologically more likely to respond to radioligand therapy. This may explain the higher PSA response rates and improved imaging outcomes observed in such studies. In this context, PSMA PET is not just a tool to verify target presence; it plays a central role in stratifying patients, optimizing treatment allocation and improving the balance between the efficacy and toxicity of PSMA-targeted therapies.
Advances in PET scanners
In recent years, significant technological development in PET scanning has led to the emergence of digital PET scanners that significantly outperform traditional analog scanners, with improved sensitivity, resolution and quantification capabilities, particularly for detecting smaller lesions.
The distinction between digital and analog PET systems has important clinical implications. One of the most significant innovations is the introduction of the next-generation solid-state digital photon counting (DPC) PET/CT system. These scanners demonstrated significant improvements in spatial and temporal resolution, increased sensitivity and superior count-rate performance.44 The key breakthrough is the use of DPC detectors, which eliminate the analog conversion process through direct digitization of photon signals. This allows for more accurate photon detection, which leads to better energy and timing resolution that is crucial for improved time-of-flight (TOF) imaging. These advancements contribute to sharper images, faster scans and reduced radiotracer doses, thereby improving both diagnostic accuracy and patient safety.
Another important advantage of digital PET scanners is the improvement in the signal-to-noise ratio (SNR) compared with traditional analog PET systems. A head-to-head comparison of human and phantom images demonstrated that digital PET scanners provide significantly higher image quality.45 This is especially evident in small lesion detectability, where digital systems show superior contrast recovery and improved quantitative accuracy.
More recently, a prospective study compared the performance of digital and analog 68Ga-PSMA-11 PET/CT in detecting post-prostatectomy biochemical recurrence in patients with prostate cancer. Despite demonstrating comparable lesion detection rate (71.8% vs 74.4%), sensitivity (85.0% vs 90.0%) and PPV (both 100.0%), the digital PET/CT system detected more lesions (139 vs 111) and had higher SUVmax (14.3 vs 10.3) and higher kappa index (0.657 vs 0.502) than analog PET/CT, regardless of serum PSA levels.46
PSMA radiopharmaceuticals for the treatment of prostate cancer: 177Lu-PSMA-617 and 177Lu-PSMA-I&T
In targeted radionuclide therapy for prostate cancer, the discussed interchangeability between the two primary radioligand therapies, 177Lu-PSMA-617 and 177Lu-PSMA-I&T (also known in a separate formulation as 177Lu-PNT2002), allows for greater flexibility in treatment selection based on availability, cost and patient-specific responses to therapy (Table 2).47 However, 177Lu-PSMA-617 is currently the only therapy with regulatory approval, supported by gold standard phase III clinical trials, such as the VISION and PSMAfore trials.35,48–50 Both 177Lu-PSMA-617 and 177Lu-PSMA-I&T have been extensively used over the last decade, especially in Europe, including significant off-trial use of 177Lu-PSMA-I&T, particularly due to lack of access to 177Lu-PSMA-617 in certain regions.
In a clinical setting, 177Lu-PSMA-I&T has been assessed in the phase III SPLASH trial, demonstrating efficacy and safety in patients with metastatic castration-resistant prostate cancer (mCRPC) progressing on an androgen receptor pathway inhibitor (ARPI) (Table 2).51,52 In this study, 412 patients with PSMA-expressing mCRPC were randomized 2:1 to receive either 177Lu-PSMA-I&T (6.8 GBq every eight weeks for up to four cycles) or an ARPI switch (abiraterone/enzalutamide). The trial met its primary endpoint, showing a median radiographic progression-free survival (rPFS) of 9.5 months with 177Lu-PSMA-I&T compared with 6.0 months with ARPI (HR: 0.71 [95% CI: 0.55–0.92]; p=0.0088) (Figure 3). This benefit was maintained across key prespecified subgroups. Furthermore, 38.1% of patients in the 177Lu-PSMA-I&T arm achieved an overall response, compared with 12.0% of patients in the ARPI arm (p=0.0021), with a median duration of response (DoR) of 9.4 months versus 7.3 months, respectively. A PSA decline of ≥50% was observed in 35.7% of patients treated with 177Lu-PSMA-I&T and 14.6% of those treated with ARPI.
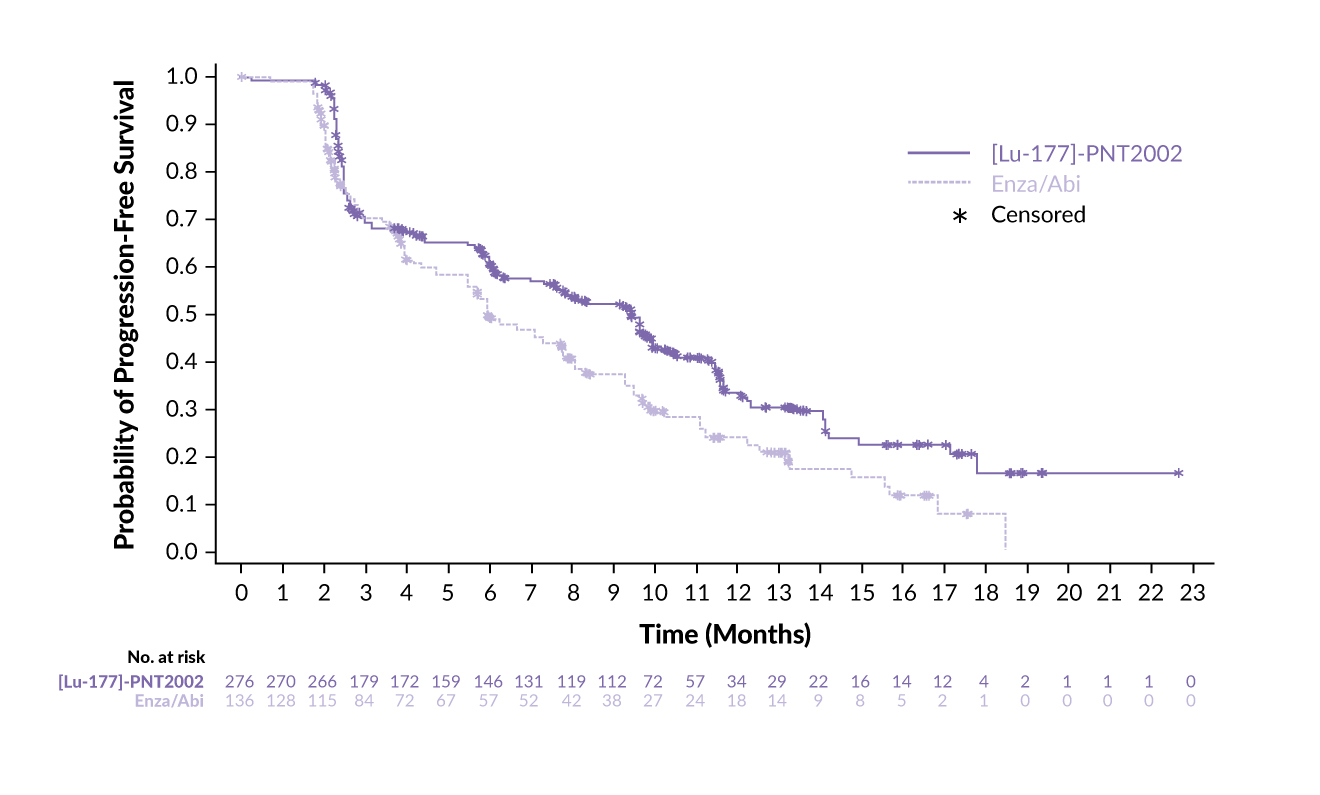
Promising outcomes were also reported in the phase III ECLIPSE trial, which assessed 177Lu-PSMA-I&T versus ARPI switch (abiraterone or enzalutamide) in patients with mCRPC who had been previously treated with ARPI and had not received prior taxane therapy.53,54 The study demonstrated a statistically significant and clinically meaningful improvement in the median rPFS with 177Lu-PSMA-I&T compared with ARPI,54 but details have not been communicated yet. Beyond clinical trials, real-world studies have further demonstrated the clinical activity and tolerability of 177Lu-PSMA-I&T in patients with mCRPC treated in routine practice.55,56
Two recent prospective dosimetry studies provide important insights into the organ-specific radiation exposure of 177Lu-PSMA-617 in the VISION trial (7.4 GBq per cycle for up to six cycles) and 177Lu-PSMA I&T in the SPLASH trial (6.8 GBq per cycle for up to four cycles) (Table 2). Although both agents demonstrated favorable tumor uptake, important differences in normal organ dosimetry, particularly kidney exposure, likely influenced the distinct dosing regimens used in these trials. In the VISION study, 177Lu-PSMA-617 showed mean absorbed doses per GBq of 0.43 Gy in the kidneys, 2.10 Gy in the lacrimal glands, 0.63 Gy in salivary glands and 0.035 Gy in red marrow.57 The cumulative kidney dose after six cycles was 15 Gy, well below the traditional 23 Gy threshold derived from external beam radiation therapy (EBRT). In contrast, 177Lu-PSMA I&T was associated with a higher mean renal absorbed dose of 0.73 Gy/GBq, with cumulative estimates exceeding 23 Gy in 22.2% of patients, potentially due to both higher physiological uptake and methodological limitations of the dosimetry studies, such as heterogeneous imaging protocols, fewer post-therapy time points and less frequent use of late imaging, all of which can lead to overestimation of renal dose.42 Tumor absorbed doses were broadly similar between the agents. Differences in kidney exposure, despite the lack of evidence for an association with kidney function, influenced the FDA-mandated dosing regimens. These data suggest that 177Lu-PSMA-617 may offer greater flexibility for prolonged or intensified dosing compared to 177Lu-PSMA I&T because of its more favorable renal dosimetry profile.
Studies directly comparing 177Lu-PSMA-617 and 177Lu-PSMA-I&T suggest mutual noninferiority in the treatment of mCRPC (Table 2). A systematic review and meta-analysis of 24 studies involving 1,192 heavily pretreated patients who received 177Lu-based therapies found no significant differences in PSA decline ≥50% or grade 3−4 toxicities between the two radioligands.58 More specifically, 44% of patients treated with 177Lu-PSMA-617 and 36% of those treated with 177Lu-PSMA-I&T achieved a PSA decline of ≥50%. Both therapies were well tolerated, with low rates of grade 3−4 toxicity, the most common being anemia (8%). These results, supported by an updated analysis of this study58 and a more recent meta-analysis,59 suggest that both radiopharmaceuticals can be considered radioequivalent in terms of efficacy and safety for the treatment of mCRPC.
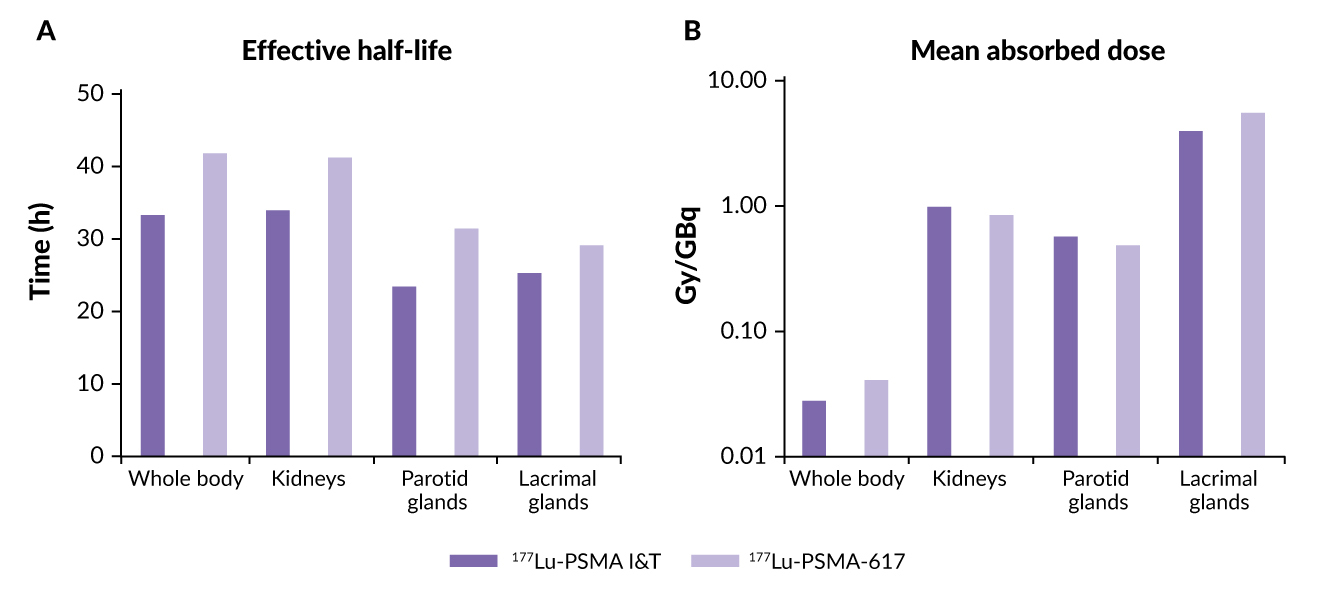
A more recent comparative dosimetry study between 177Lu-PSMA-I&T and 177Lu-PSMA-617 indicated minor differences between the two agents (Table 2). These included a shorter whole-body half-life for 177Lu-PSMA I&T than for 177Lu-PSMA-617 (35 vs 42 hours) and a significantly higher mean whole-body dose for 177Lu-PSMA-617 compared with 177Lu-PSMA I&T (p<0.00001) (Figure 4).60 Results also showed that the renal dose was significantly lower for 177Lu-PSMA-617 than for 177Lu-PSMA I&T (p=0.0015), while parotid gland doses were comparable (p=0.27). Tumor metastases exhibited higher initial uptake with 177Lu-PSMA I&T versus 177Lu-PSMA-617 but had a shorter tumor half-life (p<0.00001). Despite these differences, mean absorbed tumor doses were comparable between the two molecules (p=0.96).
The relationship between radiation dosimetry, pretherapeutic imaging and treatment outcomes of 177LuPSMA-617 was further investigated in the prospective LuPSMA trial.61 This study showed a significant association between whole-body tumor dose and PSA response at 12 weeks, with a median dose of 14.1 Gy in patients achieving a PSA decline of ≥50% versus 9.6 Gy for those achieving a PSA decline of <50% (p<0.01). There was also a significant correlation between SUVmean of whole-body tumors on 68Ga-PSMA PET and SUVmean whole-body tumor dose (p<0.001). The highest doses were observed in the salivary glands, lacrimal glands and kidneys. While SUVmean was selected as a global, reproducible parameter for uptake quantification, it should be noted that other metrics including SUVmax, SUVpeak or volumetric measures such as total lesion PSMA uptake may provide complementary insights into tumor heterogeneity and treatment-dose relationships.
A detailed tumor dosimetry analysis for 177Lu-PSMA-617 was performed in a subgroup of 29 non-randomized patients from the VISION trial who received up to six cycles of 177Lu-PSMA-617 (7.4 GBq every six weeks) in combination with standard of care (SoC).62 Single-photon emission computed tomography (SPECT)/CT scans were used to assess tumor uptake and calculate radiation-absorbed doses over time. Data from 60 delineated prostate cancer lesions were analyzed across cycles 1–6 in 18 patients with evaluable tumors in cycles 2–6. The mean absorbed dose across all tumors declined from 7.9 Gy/GBq in cycle 1 to 1.6 Gy/GBq by cycle 6, indicating a progressive reduction in radiation delivery with successive cycles. Specifically, mean absorbed doses for bone metastases decreased from 6.4 Gy/GBq to 1.3 Gy/GBq, while doses for lymphatic metastases declined from 11 Gy/GBq to 3.2 Gy/GBq. Across the six cycles, patients received a median cumulative tumor-absorbed dose of approximately 100 Gy. This trend in declining absorbed doses over time in this dosimetry substudy is consistent with the antitumor activity demonstrated by 177Lu-PSMA-617 in the VISION trial.
With the regulatory approvals of radiopharmaceuticals in several countries worldwide, the number of patients requiring treatment and consequently the number of theranostic centers, is expected to increase significantly. This expansion underscores the need for standardization and harmonization of practices across these centers to ensure consistent and high-quality patient care. A recent international survey assessing operational variations among 177Lu-PSMA treatment centers revealed notable interinstitutional discrepancies in several aspects of 177Lu-PSMA radionuclide therapy, particularly in patient selection and response assessment strategy.63 One key finding was the variation in PSMA PET eligibility criteria for 177Lu-PSMA-617. The survey showed that 68Ga-PSMA-11 was used to determine eligibility in 77% of centers, while additional pre-therapy imaging included 18F-FDG PET/CT (49%), CT (32%), renal scintigraphy (30%) and bone scintigraphy (15%) among the 84 centers for clinical standard of care, compassionate care or local research protocols. In contrast, among 42 centers for industry-sponsored trials, the proportions differed, with 26% using 68Ga-PSMA-11, 60% using 18F-FDG PET/CT, 21% using renal scintigraphy and 67% using bone scintigraphy. The survey also found variability in PSMA PET eligibility criteria across institutions. While 33% of centers relied on subjective qualitative assessment of PSMA positivity, 23% applied VISION criteria and 13% applied TheraP criteria. Regarding imaging response criteria, 35% of centers used PSMA PET progression (PPP) criteria, 33% had no standardized criteria, 23% used Response Evaluation Criteria in Solid Tumors (RECIST) and 21% followed Prostate Cancer Clinical Trials Working Group 3 (PCWG3) criteria, among others.
Ongoing studies of α-emitting radiopharmaceuticals
Compounds emitting α particles, such as Actinium-225 (225Ac) and Plumbum-212 (212Pb), are emerging as promising agents in targeted radiotherapy due to their unique radiation properties.64 Compared with β-emitters, which release lower-energy electrons over a longer range, α-emitters are much more energetic and therefore induce a greater number of double-strand DNA breaks in tumor cells.
Several novel 225Ac-PSMA-containing therapies are currently under investigation in advanced clinical phases. The phase I AcTION study was designed to assess the safety of 225Ac-PSMA-617 in men with PSMA-positive prostate cancer, regardless of prior 177Lu-PSMA-617 radioligand therapy.65 This open-label, dose-escalation study aimed to assess safety outcomes over approximately 18–24 months, with a total study duration of 48 months. Patients may receive up to six treatment cycles, with dose modifications permitted. The primary endpoints are safety and dose-limiting toxicity (DLT), while secondary endpoints include response rates, PFS and overall survival (OS).
PSMAcTION is a phase II/III, open-label, international, multicenter, randomized study evaluating 225Ac-PSMA-617 (AAA817) versus SoC in adult patients with PSMA-positive mCRPC who had received prior treatments with ARPI and taxane-based chemotherapy and progressed on or after 177Lu-PSMA-617 targeted therapy.66 The study consists of two parts: a phase II part designed to collect additional information supporting the proposed dose of 225Ac-PSMA-617 and a phase III part aimed to evaluate the efficacy and safety of the selected dose of 225Ac-PSMA-617 compared with the investigator’s choice of SoC. The drug will be administered in 8-week cycles (dose A and dose B). The primary endpoints in phase II part of the study are the biochemical response rate and safety/tolerability, while rPFS and OS are the primary endpoints of the phase III part.
Another major trial, AcTFirst, is a phase III study evaluating 225Ac-PSMA-617 alone or in combination with ARPI (enzalutamide/abiraterone) compared with SoC in patients with PSMA-positive mCRPC.67 Adult patients will be randomized into three treatment arms to receive either up to six doses of 225Ac-PSMA-617 (10 Mbq intravenously) plus ARPI (enzalutamide/abiraterone), 225Ac-PSMA-617 alone or standard treatment with enzalutamide/abiraterone or taxane-based chemotherapy. Crossover between the three treatment arms is not allowed. The primary endpoint is rPFS, with key secondary endpoints including OS and rPFS as determined by PSMA PET/CT imaging.
Furthermore, 225Ac-PSMA-R2 is currently being evaluated in the SatisfACtion study in patients with PSMA-positive mCRPC who had previously undergone ARPI and taxane therapy.68 In this phase I/II study, patients receive intravenous 225Ac-PSMA-R2 plus SoC once every six weeks for up to six cycles. The dose-escalation phase assesses safety, tolerability and recommended dose for expansion of 225Ac-PSMA-R2. In the dose-expansion phase, the primary endpoints are overall response rate (ORR) and PSA50 response rate. Secondary endpoints across both phases include response rates, rPFS, PFS, OS and time to symptomatic skeletal events, among others.
Another promising radiopharmaceutical, 225Ac-PSMA-I&T (FPI-2265) has shown strong clinical activity in patients with mCRPC across multiple studies.69,70 It is currently being studied in the ongoing, open-label, single-arm TATCIST study in patients with PSMA-positive, progressive disease who had received at least one prior ARPI.71 Four doses of 225Ac-PSMA-I&T at 100 kBq/kg (±10%) were administered in 8-week intervals. Patients with skeletal metastases presenting as a superscan were excluded. The recently presented initial results demonstrated that PSA50 was achieved in half of the patients (10/20) irrespective of prior treatment with Lu-PSMA radioligand therapy (Figure 5). Data further showed that in patients with a baseline PSMA SUVmean >6 (n=13), the PSA50 rate was 69%, with a durable PSA response by week 24. Safety and tolerability were consistent with other published studies of 225Ac-PSMA radioligand therapies. The majority of treatment-related adverse events (TRAEs) were grade 1–2, with xerostomia being the most common event.
_change_from_baseline_during_the_t.jpg)
225Ac-PSMA-I&T therapy is also currently being assessed in the investigator-initiated phase I, single-center 225Ac-PSMA-Imaging & Therapy (I&T) trial, which aims to assess the safety and tolerability of 225Ac-PSMA-I&T to determine the optimal dose for the phase II trial.72 This open-label study follows a repeated dose-escalation and expansion design in patients with PSMA-positive mCRPC who have undergone at least one line of chemotherapy and/or one line of nonsteroidal antiandrogen. Patients will be treated with increasing levels of 225Ac-PSMA-I&T activity per cycle, following an accelerated 3 + 3 design, which allows enrollment of the next dose-level cohort in the absence of dose-limiting toxicity, even as the previous cohort continues. Up to four treatment cohorts will be explored, including three dose-escalation cohorts and one expansion cohort at the recommended dose. A maximum of 30 patients will be enrolled, all of whom will be evaluated for safety. Dosimetry has also been performed following the first administration of 225Ac-PSMA-I&T in the dose-escalation phase.
212Pb-ADVC001 is a novel PSMA-targeted radioligand labeled with the α-emitting isotope Plumbum-212, which is being investigated in the ongoing, prospective phase I/II TheraPb study in patients with mCRPC.73 During phase Ib, escalating doses of 212Pb-ADVC001 are administered every six, four or two weeks using an i3+3 design in patients with prior exposure to at least one ARPI and taxane-based chemotherapy, with backfilling of up to 20 patients per cohort to further evaluate safety, tolerability and early efficacy. Phase IIa will assess the recommended phase II dose across three groups: those previously treated with ARPI but not taxanes, those treated with both ARPI and taxanes for mCRPC and those previously exposed to 177Lu-PSMA.
Other PSMA-based radioligands
Therapies based on PSMA-617 and PSMA I&T require high doses of these radioligands due to their rapid blood clearance, potentially resulting in systemic toxicity. To prolong circulation time, a novel PSMA-targeted radioligand, ¹⁷⁷Lu-EB-PSMA, was developed by conjugating 177Lu-PSMA-617 with a truncated Evans Blue (EB) moiety and a DOTA chelator.74 The EB component binds to plasma albumin, thereby delaying systemic clearance, enhancing tumor uptake and reducing the required radioligand dose. Tumor accumulation of 177Lu-EB-PSMA was found to be approximately three times higher than that of 177Lu-PSMA-617.
In a dose-escalation study, administration of 2.12 ± 0.19 GBq per dose of 177Lu-EB-PSMA demonstrated promising efficacy and an acceptable safety profile.75 Its clinical utility was further evaluated in a single-arm, low-dose, prospective phase I study that enrolled 30 patients with progressive mCRPC previously treated with taxane-based chemotherapy and second-generation androgen deprivation therapy (ADT).76 Patients received up to three cycles of approximately 2.0 GBq per cycle at 8-week intervals. The primary endpoint was safety and the additional primary endpoint was therapeutic efficacy measured by PSA and molecular imaging responses. The secondary endpoints included PSA PFS, OS and patient-reported health-related quality of life (HRQoL). The study reported grade 3 hematologic adverse events (AEs) in 33.3% of patients.76 A PSA decline of ≥50% was observed in 56.7% of patients and the median PSA PFS and OS were 4.6 and 12.6 months, respectively. HRQoL scores improved significantly following therapy. These findings suggest that administering 2.0 GBq of 177Lu-EB-PSMA for up to three cycles yields a therapeutic response and hematologic toxicity profile comparable to 7.4-GBq regimens of 177Lu-PSMA-617 across 4–6 cycles, positioning 177Lu-EB-PSMA as a promising alternative for mCRPC treatment.
TLX591 (177Lu-DOTA-rosopatamab) is another 177Lu-labeled, DOTA chelator-conjugated monoclonal antibody, which demonstrated high tumor specificity, low off-target toxicity and prolonged tumor retention, with a favorable safety profile.77 Based on early-phase studies, including the phase I ProstACT SELECT trial, showing consistent uptake between TLX591 and 68Ga-PSMA-11 imaging, the multinational, open-label, randomized phase III ProstACT GLOBAL trial was designed to investigate TLX591 in 400 patients with PSMA-positive mCRPC who have progressed following prior ARPI therapy. Patients are randomized 2:1 to receive protocol-defined standard of care (ARPI or docetaxel) with or without two intravenous doses of TLX591 (2.8 GBq, 14 days apart). The primary endpoint is rPFS, with secondary endpoints including 5-year OS, tumor response rate, time to symptomatic skeletal events, QoL and safety profiles.
Preclinical studies have demonstrated synergistic antitumor activity between PSMA-targeted radioligand therapy and ARPI enzalutamide. The multicenter, randomized, phase II ARROW trial therefore aimed to evaluate the efficacy and safety of Iodine-131 (131I)-PSMA-1095 plus enzalutamide versus enzalutamide monotherapy in chemotherapy-naïve mCRPC patients with disease progression on abiraterone and positive 18F-DCFPyL PET/CT.78 A total of 120 men ineligible for or refusing taxane-based chemotherapy were randomized 2:1 to receive 131I-PSMA-1095 plus enzalutamide or enzalutamide alone. The primary endpoint of PSA50 response rate was met, with a statistically significant PSA50 improvement in the combination arm compared with the enzalutamide monotherapy arm (63% vs 31%; p=0.003). The median rPFS favored the combination therapy, although the difference was not statistically significant (14 months vs 11.5 months; p=0.10). The median OS was 18.8 months with 131I-PSMA-1095 plus enzalutamide versus 22.0 months with enzalutamide monotherapy (p=0.59). The safety profile was consistent with that of other drugs of this type, with grade ≥3 treatment-emergent AEs occurring in 66% and 41% of patients in the combination and monotherapy arms, respectively. Importantly, the use of 131I as the β-emitting isotope may impact both efficacy and toxicity relative to 177Lu-labeled PSMA ligands. 131I emits higher-energy β-particles with a longer penetration range, potentially enhancing crossfire effects in bulky or heterogeneous tumors but also increasing the risk of off-target irradiation.79 In addition, the high-energy γ-emission of 131I enables imaging and dosimetry but contributes to greater whole-body radiation exposure and requires stricter radiation safety precautions compared with 177Lu.
Future outlook and conclusions
As theranostics evolves, new ligands, including antibodies, have emerged as potential options.80 However, in the ongoing comparison between small-molecule peptides and antibodies regarding targeting efficiency and toxicity profiles, small-molecule peptides remain the preferred choice as they provide superior tumor uptake, faster clearance, lower cost and fewer side effects.81 The development of new ligands and the shift toward α-emitter isotopes underscore the need for prospective, high-quality clinical data to further refine treatment strategies and improve patient outcomes in prostate cancer therapy.
In the dynamic field of PSMA PET/CT imaging for prostate cancer, the selection of radiotracer largely relies on availability. Currently, 68Ga-PSMA-11 and 18F-DCFPyL are considered comparable in performance, making them the most widely used options. The recently approved radiotracer, 18F-rhPSMA-7.3 appears to offer similar efficacy but clinical experience remains limited. In contrast, caution is advised with 18F-PSMA-1007 due to its problematic benign bone uptake, which can lead to false-positive findings.
For therapeutic applications, there are currently no head-to-head prospective studies comparing 177Lu-PSMA-617 and 177Lu-PSMA-I&T. Retrospective analysis demonstrated differences in normal organ distribution between the two agents, with similar mean absorbed tumor doses, efficacy and safety profiles in the treatment of mCRPC. The first prospective dosimetry data from the phase III VISION and SPLASH trials indicate that renal absorbed doses will dictate how these radiopharmaceuticals can be effectively dosed while maintaining a favorable risk/benefit ratio. Until proven otherwise, the recommended dosing of 177Lu-PSMA-617 is up to six cycles, six weeks apart at 7.4 GBq. Beyond radiotracer selection, patient selection, reader expertise and technical considerations play a key role in optimizing both diagnosis and treatment outcomes for patients with advanced prostate cancer.
Conflict of interest
Ken Herrmann reported receiving consultant fees from Advanced Accelerator Applications (a Novartis company), Amgen, AstraZeneca, Bain Capital, Bayer, Boston Scientific, Convergent, Curium, Debiopharm, EcoR1, Fusion, GE Healthcare, Immedica, Isotopen Technologien München, Janssen, Merck, Molecular Partners, NVision, POINT Biopharma, Pfizer, Radiopharm Theranostics, Rhine Pharma, Siemens Healthineers, Sofie Biosciences, Telix and Theragnostics and ymabs; receiving research grants from Advanced Accelerator Applications, Boston Scientific and Janssen; having stock or other ownership interests with AdvanCell, Aktis Oncology, Convergent, NVision, Pharma 15 and Sofie Biosciences. Wolfgang P. Fendler reported fees from SOFIE Bioscience (research funding), Janssen (consultant and speaker), Perceptive (consultant and image review), Bayer (consultant, speaker and research funding), Novartis (speaker and consultant), Telix (speaker), GE Healthcare (speaker and consultant), Eczacıbaşı Monrol (speaker), Abx (speaker), Amgen (speaker and research funding), Urotrials (speaker), Lilly (consultant) and AstraZeneca (research funding) outside of the submitted work. Marco Cuzzocrea reported receiving honoraria from Novartis and travel support from Novartis and BE-imaging. These funding entities did not play a role in the development of the manuscript and did not influence its content in any way. Gaetano Paone declared that the manuscript was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was partly funded by Prostate Cancer Foundation TACTICAL Award No 22TACT01, Thera4Care by the Innovative Health Initiative Joint Undertaking (IHI JU) under grant agreement No 101172788 and ILLUMINATE by the Innovative Health Initiative Joint Undertaking (IHI JU) under grant agreement No 101172722.
Author contributions
All authors contributed to and approved the final manuscript.