

Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by clonal proliferation of plasma cells in the bone marrow, leading to a range of complications such as osteolytic bone lesions, anemia, hypercalcemia and renal impairment. It accounts for approximately 10% of all hematologic cancers, with a rate of new cases of 7.2 per 100,000 individuals per year.1,2 Despite significant advancements in treatment, MM remains incurable in most cases. MM is a highly complex and heterogeneous disease, characterized by high genetic variability and clonal evolution, both at diagnosis and relapse.3,4 The presence of certain high-risk cytogenetic abnormalities in MM, including 17p del, 1q amplification, t(14;16), t(14;20) and t(4;14), is associated with more aggressive disease, poor prognosis and increased resistance to standard therapies. Over time, MM can evolve under therapeutic pressure, leading to clonal selection and the emergence of resistant subclones,5 with increased mutational burden and immune exhaustion contributing to treatment resistance, reducing immune function and further impacting therapeutic efficacy. Other treatment effects affecting long-term outcomes include infectious complications and myelosuppression resulting from immune compromise and bone marrow suppression, end-organ injury (renal, cardiac, skeletal, pulmonary, vascular and neural) and extramedullary “escape” indicating more aggressive, difficult-to-treat disease. Together, this underscores the critical need to develop novel therapeutic approaches to improve patient outcomes in both newly diagnosed and relapsed settings.
Alkylating agents are a foundational class of drugs in MM therapy that exert their anticancer effects by inducing DNA cross-linking and subsequent cell cycle arrest, thus disrupting replication and inducing apoptosis.6 Classic alkylating agents, such as melphalan and cyclophosphamide, have long been integral to MM therapy, particularly in transplant-ineligible patients and combination regimens. Recently, melphalan flufenamide (melflufen), a novel peptide-conjugated alkylating agent, has shown promise in MM.7 Melflufen delivers melphalan directly into myeloma cells through aminopeptidase activity, achieving higher intracellular concentrations with potentially reduced toxicity. This targeted mechanism offers a unique therapeutic option for patients with relapsed or refractory MM, including those resistant to conventional alkylating agents. In this review, we discuss current therapeutic options for MM, with a particular focus on alkylating agents, specifically melflufen. We delineate its mechanisms of action, data from preclinical studies and findings from clinical trials in patients with MM.
Evolution of therapeutic approaches and current treatment landscape in MM
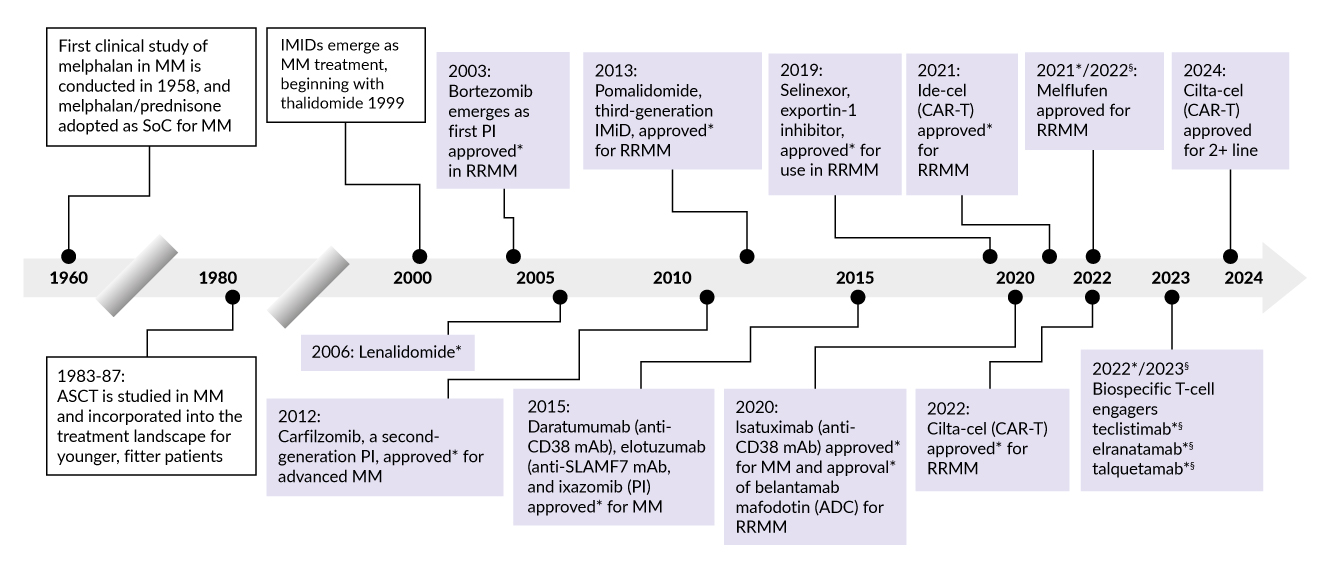
Over the past 24 years, the therapeutic landscape of MM has been transformed through a series of innovative treatments (Figure 1). The use of autologous stem cell transplantation (ASCT), which began in the 1980s, established a foundational strategy for treating younger, fit MM patients, offering substantial survival benefits. The emergence of immunomodulatory drugs (IMiDs) began with thalidomide in 1999, adding a novel class of agents that modulates the immune environment to suppress tumor growth.8 In 2003, bortezomib was approved as the first proteasome inhibitor (PI), a drug class that inhibits protein degradation pathways essential for MM cell survival, particularly in relapsed/refractory MM (RRMM).9 Subsequent approvals have introduced second- and third-generation IMiDs, such as lenalidomide (2006) and pomalidomide (2013), enhancing efficacy for patients with resistant disease. The development of monoclonal antibodies (mAbs), including daratumumab and elotuzumab in 2015, marked another milestone by specifically targeting antigens like CD38 on MM cells, improving responses in combination regimens.10 Recent advancements include B-cell maturation antigen (BCMA)-targeting chimeric antigen receptor (CAR) T-cell therapies idecabtagene vicleucel (ide-cel) and ciltacabtagene-autoleucel (cilta-cel) approved for RRMM in 2021 and 2022, respectively.11 Several bispecific T-cell engagers (BiTEs) such as teclistimab, elranatamab and talquetamab bridging MM cells and T cells to enhance cytotoxicity have been approved in the last two years.12
__focus_on_the_past_24_years.jpg)
Furthermore, various other agents and combination regimens are currently approved or under investigation, including cereblon E3 ligase modulators (CELMoDs), antibody-drug conjugate (ADC) belantamab mafodotin, targeted therapies such as venetoclax, selinexor and melflufen, CAR natural killer (NK) cell therapies, immune checkpoint inhibitors (ICIs) and immunocytokines (Figure 2). These innovative treatments underscore a shift towards highly targeted and individualized approaches in MM that address specific molecular and immunological aspects of MM pathology.
_in_2025__therapies_approved_or_under_investigation.jpeg)
Alkylating agents in treatment of MM
Classical alkylating agents in hematologic malignancy treatment
The use of alkylating agents for the treatment of hematologic malignancies dates back to the seminal study by Goodman and Gilman in the 1940s which set the foundation for the use of alkylating agents in oncology by demonstrating the efficacy of nitrogen mustard compounds, such as methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride in treating Hodgkin’s disease, lymphosarcoma and leukemia.15 Cyclophosphamide16 received initial approval from the U.S. Food and Drug Administration (FDA) in 1959; subsequently, in 1964, based on the research conducted by Bergsagel et al.,17 approval was granted to melphalan, which has become a fundamental component in the treatment of MM and the backbone for high-dose chemotherapy in combination with ASCT. The initial exploration of high-dose melphalan (HDM) in MM began in the late 1970s, with pioneering work by McElwain and Powles, who investigated high-dose strategies to overcome resistance in plasma cell malignancies. Published in The Lancet, their research demonstrated promising outcomes in patients with plasma cell leukemia and MM,18 leading to the use of HDM as a standard pre-transplant conditioning regimen. Subsequent studies further confirmed the effectiveness of HDM when combined with ASCT, showing significant improvements in progression-free survival (PFS) and overall survival (OS) in MM patients.19–21 The clinical benefits of HDM-ASCT have persisted even with the advent of novel agents, affirming its role in the modern treatment landscape.
Combination regimens incorporating alkylating agents: Where are we today?
The integration of classical alkylating agents, such as cyclophosphamide, melphalan and bendamustine, in combination regimens with novel therapies, including PIs, IMiDs and mAbs, has provided clinical benefits across various patient populations. The guidelines of the International Myeloma Working Group (IMWG) recommend HDM plus ASCT as frontline therapy, with the addition of bortezomib, melphalan and prednisone (VMP) at first relapse.14 Similarly, the European Society for Medical Oncology (ESMO) guidelines recommend HDM plus ASCT as frontline therapy, as well as daratumumab plus VMP (Dara-VMP) or VMP alone as initial treatment options.22 Cyclophosphamide is considered as an alternative to melphalan in combination with bortezomib and dexamethasone (VCd) at first relapse14 and in combination with pomalidomide and dexamethasone (PCd)14,22 or bortezomib, dexamethasone, thalidomide, cisplatin, doxorubicin and etoposide (VdT-PACE)14 at second or higher relapses. Bendamustine is currently not included in the guidelines.
The efficacy of combination regimens containing melphalan, cyclophosphamide and bendamustine has been evaluated in several clinical trials. In the phase III ALCYONE study, Dara-VMP significantly improved OS in patients with newly diagnosed MM (NDMM) (n=706), with 3-year OS rate of 78.0% versus 67.9% with VMP alone (HR: 0.60 [95% CI: 0.46–0.80]; p=0.0003).23 The objective response rate (ORR) was 90.9% versus 73.9% and the median PFS was 36.4 months versus 19.3 months (HR: 0.42), respectively. The addition of cyclophosphamide to Pd achieved the ORR of 70.6% versus 47.1% with Pd alone in patients with RRMM (n=102) in the phase II MUKseven study.24 The median PFS was 6.9 months versus 4.6 months, respectively (which was not, however, significantly different as per pre-defined criteria). Yet, in a phase III study VCd and Vd-alone regimens demonstrated similar efficacy in RRMM patients (n=96) after a median follow-up of 24 months, with a median time to disease progression (TTP) of 9.9 months in the VCd arm versus 12.6 months in the Vd arm (p=0.192) and ORR of 70% versus 74%, respectively.25 Bendamustine and prednisone (BP) was superior to standard melphalan and prednisone (MP) in previously untreated MM patients (n=131) in a phase III study with respect to ORR (75% vs 70%), time to treatment failure, cycles needed to achieve maximum remission and quality of life (QoL).26
The combination chemotherapy regimen of dexamethasone, cyclophosphamide, etoposide and cisplatin (DCEP) is utilized as a salvage therapy for RRMM patients,27,28 with the aim to control disease during aggressive progression27,29,30 and as a bridge to ongoing therapies.27,31 DCEP has been demonstrated to be an effective and safe third-line salvage treatment in a cohort of 12 patients with RMMM,29 with an ORR of 58.3%, a median duration of response (DoR) of nine months, and four patients successfully proceeding to peripheral blood stem cell mobilization and transplant. In a study comparing three salvage chemotherapy regimens (DCEP group, n=52), the median PFS was 3.8 months, the median OS was 8.9 months and ORR was of 52%.27 The study by Yuen et al. further supported DCEP as an effective bridge to definitive therapy in a study involving 65 patients, showing an ORR of 55% and an OS of 9.6 months.31 For patients who were bridged to ASCT, the median OS was extended to 32.8 months compared with 10.7 months for those who were not. However, DCEP is associated with hematological toxicities, primarily with grade ≥3 neutropenia, anemia and thrombocytopenia, as well as mucositis, alopecia and peripheral neuropathy.31 Sepsis-related treatment mortality (TRM) is a significant risk with DCEP; therefore, concurrent use of prophylactic granulocyte colony-stimulating factor (G-CSF) is recommended.27,31
Potential toxicities of alkylating agents
Treatment using alkylating agents is associated with a broad range of potential toxicities. Hematologic toxicities are particularly common, with neutropenia, thrombocytopenia and anemia being the most prevalent32–34; other toxicities include gastrointestinal adverse events (AEs)32,34 including nausea, vomiting, mucositis and infections, and alopecia.32 Furthermore, HDM followed by ASCT is associated with an increased risk of long-term health consequences in MM survivors, including cardiovascular disease, endocrine dysfunction and ocular AEs. Among 630 MM patients who survived ≥2 years after HDM-ASCT, 40% had higher odds of developing grade 3–4 chronic health conditions compared with siblings, with 10-year cumulative incidence of 57.6%.35 Approximately one-third of long-term survivors post-ASCT had clinically significant distress based on patient reported outcomes.36
Alkylating agents may also potentiate the risk of secondary malignancies, such as myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), which have been linked to prolonged exposure to alkylating agents and genotoxic effects.34,37,38 Whole-genome sequencing (WGS) has demonstrated a significant increase in mutational burden in residual MM cells after HDM and ASCT. Although the number of mutations at diagnosis was similar between patients receiving lenalidomide plus bortezomib and dexamethasone (RVd) alone and those receiving RVd plus HDM, the HDM group had significantly more mutations at relapse (9,242 vs 13,383; p=0.005). The increased incidence of secondary AML and MDS following HDM-ASCT was highlighted by the data from the Center for International Blood and Marrow Transplant Research (CIBMTR) involving 9,028 ASCT recipients.39 While according to the Surveillance, Epidemiology and End Results (SEER) data, the risks for AML/MDS in Hodgkin lymphoma (HL), non-Hodgkin lymphoma (NHL) and plasma cell myeloma patients were 5–10 times the background rate, relative risks were 10–50 for AML and approximately 100 for MDS in the autotransplant cohort. The mutation accumulation rate was 4.1-fold higher in the RVd plus HDM group compared with the RVd-alone group (158.3 vs 38.3 mutations/month; p=0.003). WGS analysis of 40 cases of therapy-related myeloid neoplasms (tMN) across patients with MM, NHL and solid tumors revealed an elevated single-base substitution mutational burden associated with HDM exposure.40 Among the proposed mechanisms underlying tMN development is the acquisition of the SBS-MM1 mutational signature shown to be caused by melphalan exposure,38,41–44 and the chemotherapy-induced escape and reinfusion of pre-leukemic clones during leukapheresis. Prolonged cytopenia and subsequent MDS were observed also in 19% of RRMM patients who underwent BCMA-directed CAR T-cell therapy, particularly after multiple ASCTs.41
Increasing rationale for deferred ASCT approach in NDMM
Given the treatment AEs and genotoxic risks linked to HDM, there is currently an increasing rationale to consider deferring HDM-ASCT, reserving it as a potential salvage therapy in selected transplant-eligible patients. Recent studies have shown that transplant-based approaches, while achieving a longer PFS than non-transplant strategies, do not confer an OS advantage. Additionally, highly active triplet and quadruplet regimens using PIs, IMiDs and mAbs result in high rates of minimal residual disease (MRD) negativity even without HDM-ASCT in the initial treatment.
The phase III DETERMINATION trial has supported this rationale by failing to demonstrate the OS benefit of RVd plus ASCT over RVd alone in patients with NDMM.45 In this study, adult patients with symptomatic MM received one cycle of RVd and then were randomized in a 1:1 ratio to receive two additional RVd cycles plus stem-cell mobilization followed by either five additional RVd cycles (n=357) or HDM plus ASCT followed by two additional RVd cycles (n=365). Both groups received lenalidomide until disease progression or unacceptable side effects. The primary endpoint was PFS. At a median follow-up of 76.0 months, the risk of progression was 53% higher in the RVd-alone arm compared with the transplantation arm (HR: 1.53 [95% CI: 1.23–1.91]; p<0.001), with a median PFS of 46.2 months versus 67.5 months, respectively. However, there was no statistically significant difference in OS, with 5-year OS rates of 79.2 months with RVd alone versus 80.7 months with RVd plus ASCT (HR: 1.1 [95% CI: 0.73–1.65]; p=0.99). MRD negativity rates were higher in the ASCT group (54.4% vs 39.8%), indicating a deeper initial response with transplant, which, however, did not translate into long-term survival benefits. Only 28.0% of patients in the RVd-alone arm had received ASCT at any time following the end of treatment to date, while 72% received second-generation novel therapies including PIs, IMiDs or mAbs. Furthermore, RVd plus ASCT regimen was associated with higher rates of acute toxicities, a transient but clinically meaningful decrease in QoL and an increased risk of hematologic second primary malignancies, including AML and MDS. Together, these data highlight the potential of delaying ASCT in selected patients without compromising outcomes, particularly as novel therapies continue to improve the initial treatment responses.46,47
Melflufen in MM: Mechanisms of action and preclinical evidence
Peptide-drug conjugates (PDCs) represents an innovative approach in cancer therapy, utilizing a tumor-targeting peptide to deliver a cytotoxic payload directly to cancer cells.48 This platform is designed to concentrate toxic payload by exploiting the lipophilicity of the molecule49 and high aminopeptidase activity in tumor cells,50 promoting efficient cellular uptake and sustained cytotoxicity within the tumor environment while protecting healthy cells and minimizing systemic exposure.51 Melflufen is the first-in-class alkylating PDC leveraging aminopeptidases in MM. The lipophilic structure and diffusion characteristics of melflufen facilitate its rapid entry into cells, where the cytotoxic payload is released by peptidase-driven hydrolysis and irreversibly damages tumor DNA, inducing apoptosis.49,52–54 The chemical linker in the melflufen molecule is composed of a dipeptide which can be hydrolyzed by aminopeptidases, metalloenzymes that contribute to proteolysis by catalyzing the hydrolysis of terminal amino acid residues from proteins or peptides and function downstream of the ubiquitin–proteasome pathway. Aminopeptidases generally exhibit elevated expression in RRMM compared with NDMM. Five aminopeptidase genes (ERAP2, XPNPEP1, DPP3, RNPEP and CTSV) have been found to be differentially expressed in MM; among them only the expression level of ERAP2 was decreased in RRMM versus NDMM samples.50 Elevated expression levels of specific aminopeptidase genes have significant prognostic implications in MM, correlating with more aggressive disease progression and poor survival.50 Aminopeptidases also mediate the hydrolysis of melflufen to melphalan and para-fluoro-L-phenylalanine ethyl ester which could explain higher activity of melflufen in samples from RRMM patients in vitro.50
Preclinical studies in myeloma cell lines and animal models have shown that melflufen targets both nuclear and mitochondrial DNA, which disrupts essential cellular processes and triggers cell death, resulting in the rapid induction of apoptosis and a significant concentration-dependent decrease in cell viability across various MM cell lines.55,56 Melflufen action is associated with an increase in calreticulin exposure on the cell surface (Dharminder Chauhan, Dana-Farber Cancer Institute, personal communication), enhancing its immunogenicity and potentially stimulating immune-mediated cell death. Current therapy puts clonal selection pressure on myeloma cells, resulting in the expansion of clones that are no longer responsive to previous line therapy but are still sensitive to melflufen. Melflufen retained cytotoxic efficacy in MM cell lines, mouse xenograft models and bone marrow samples resistant to melphalan, indicating its potential to overcome specific resistance mechanisms.53,57,58 In ex vivo studies on primary bone marrow samples from MM patients with high-risk features such as del(17)p and TP53 mutations, melflufen has demonstrated superior efficacy over melphalan, exhibiting greater cytotoxicity in CD138+CD38+ plasma cells, even in the presence of TP53 mutations.59 Additionally, melflufen has shown enhanced cytotoxic effects compared with melphalan in ex vivo samples from patients with amyloid light-chain (AL) amyloidosis and in amyloidogenic plasma cell lines.60 Single-cell RNA sequencing (scRNAseq) and gene set enrichment analysis (GSEA) on bone marrow samples from 24 MM patients revealed that myeloma plasma cells that are highly sensitive to melflufen exhibited downregulation in p53 signaling pathways,61 indicating that melflufen cytotoxicity may operate independently of p53, aligning with other in vitro studies that show robust activity in TP53-deficient cells. Additionally, pathways related to the DNA damage response, including BRCA1 and ATM genes, were upregulated in samples with high melflufen sensitivity.
Clinical trials investigating melflufen in RMMM
Several clinical trials have investigated melflufen in patients with MM. These include the phase I/II O-12-M1 trial62 and the pivotal phase II HORIZON study63 evaluating the efficacy and safety of melflufen in combination with dexamethasone; the randomized, phase III OCEAN study64 that compared melflufen plus dexamethasone with pomalidomide plus dexamethasone; the phase I/IIa ANCHOR trial65 evaluating melflufen-dex in combination with either daratumumab or bortezomib; and the randomized, phase III LIGHTHOUSE trial66 that assessed the combination of melflufen with daratumumab and dexamethasone versus daratumumab monotherapy.
O-12-M1
The open-label, multicenter, phase I/II O-12-M1 trial evaluated the efficacy and safety of melflufen in combination with dexamethasone in patients with RRMM aged ≥18 years who had undergone at least two prior lines of therapy (including lenalidomide and bortezomib) and were refractory to the last line of therapy (progressed on treatment or within 60 days of the last dose).62 In phase I, the maximum tolerated dose (MTD) of melflufen was determined in a cohort of 23 patients who received melflufen (15 mg, 25 mg, 40 mg, 55 mg and 70 mg) intravenously (IV) on day 1 in 21-day cycles and 40 mg oral (PO) dexamethasone weekly. Phase II commenced with a dose expansion cohort (n=58) to further assess the effectiveness and tolerability. Patients received melflufen at MTD (40 mg) either in combination with dexamethasone (n=45) or as a single agent (n=13) until progressive disease, unacceptable toxicity or completion of eight cycles of therapy. The primary endpoints of phase II were ORR and clinical benefit rate (CBR). The key secondary endpoints included PFS, OS, TTP, DoR and safety.
In the phase II part of the study, the combination of melflufen and dexamethasone demonstrated an ORR of 31%, with 11% of patients achieving a very good partial response (VGPR) and 20% reaching a partial response (PR).62 At a median follow-up of 27.9 months in the combination cohort and 17.3 months in the single-agent cohort, the median PFS was 5.7 months compared with 4.4 months, respectively. The most common grade 3–4 AEs were reversible thrombocytopenia and neutropenia (58% each). Second primary malignancies (MDS) were reported in two patients (3%) who had a prolonged history of MM and previous immunomodulatory agent and alkylator exposure. Four deaths due to AEs occurred in the combination cohort in patients who had rapid disease progression (≤30 days of the last dose of melflufen), with grade 5 AEs including pneumonia (n=2), E. coli sepsis (n=2) and neutropenia (n=1). Two deaths were considered to be possibly melflufen-related and occurred after one dose of melflufen (both due to sepsis).
HORIZON
The pivotal single-arm, open-label, multicenter, phase II HORIZON study was designed to evaluate melflufen in combination with dexamethasone in RRMM patients aged ≥18 years who had undergone at least two prior lines of therapy (including an IMiD and a PI) and were refractory to pomalidomide and/or an anti-CD38 mAb.63 The study population included 38% of patients with high-risk cytogenetics and 35% with extramedullary disease (EMD); 76% were triple-class (IMiD, PI and anti-CD38 mAb) refractory. Patients (n=157) received melflufen (40 mg IV on day 1 of each 28-day cycle) plus dexamethasone (40 mg or 20 mg in patients aged ≥75 years, PO once weekly). The primary endpoint was ORR. The secondary endpoints were DoR, PFS, OS and safety.
The study reported an ORR of 29% (including a VGPR of 11% and a stringent complete response [sCR] of 1%) and a CBR of 45% for the combination of melflufen and dexamethasone in the overall population. In the triple-class refractory population, ORR, VGPR and CBR were 26%, 11% and 39%, respectively.63 At a median follow-up of 14 months, the median PFS was 4.2 months, whereas the median OS reached 11.6 months among all treated patients. In the subset of triple-class refractory patients the median PFS and OS were 3.9 months and 11.2 months, respectively (Figure 3). The median DoR was 5.5 months in all treated patients and 7.6 months in the triple-class refractory patients.
In the subset of patients with EMD (n=55), ORR was 24% (including 25% in patients with bone-related plasmacytoma and 22% in patients with soft-tissue plasmacytoma), CBR reached 31% (including 32% in patients with bone-related plasmacytoma and 30% in patients with soft-tissue plasmacytoma) and DoR was 5.5 months.63
_and_overall_survival_(os)_in_the_horizon_study.jpg)
With respect to safety, serious AEs (SAEs) occurred in 49% of patients overall, with the most frequent being pneumonia (9%) and febrile neutropenia (5%).63 One patient developed MDS after 17 cycles of therapy in the setting of prior stem cell transplant and chemotherapy. Fatal treatment-emergent AEs (TEAEs) occurred in 10 (6%) patients, primarily due to general health deterioration associated with progressive disease and respiratory failure; none were considered to be related to melflufen.
OCEAN
OCEAN (OP-103) was a randomized, open-label, phase III study that compared melflufen plus dexamethasone (melflufen-dex) with pomalidomide plus dexamethasone (pom-dex) in patients with RRMM.64 Eligible were patients aged ≥18 years with 2–4 prior lines of therapy including lenalidomide and a PI who were refractory to lenalidomide and the last line of therapy. Stratification factors included age (<75 vs ≥75 years old), prior lines of therapy (2 vs 3–4) and the International Staging System (ISS) score (I vs II/III or I vs ≥II). Patients (n=495) were randomized 1:1 to receive 28-day cycles of either melflufen-dex (melflufen, 40 mg IV on day 1 of each cycle; dexamethasone, 40 mg PO weekly; n=246) or pom-dex (pomalidomide, PO 4 mg on days 1–21 of each cycle; dexamethasone, 40 mg PO weekly; n=249) until disease progression or unacceptable toxicity. The primary endpoint was PFS assessed by the Independent Review Committee (IRC) per the IMWG uniform response criteria. The secondary endpoints included OS, ORR and safety.
At a median follow-up of 15.5 months in the melflufen group and 16.3 months in the pomalidomide group, the study reported a PFS benefit for melflufen-dex over pom-dex, with a median PFS of 6.8 months versus 4.9 months, respectively (HR: 0.79 [95% CI: 0.64–0.98]; p=0.032), corresponding to a 21% reduction in the risk of progression or death with melflufen-dex.64,67 The ORR was 33% with melflufen-dex versus 27% with pom-dex. Yet, the OS analysis showed a negative trend for melflufen-dex in the intention-to-treat (ITT) population, with a median OS of 19.8 months versus 25.0 months in the pomalidomide group (HR: 1.14 [95% CI: 0.85–1.44]; p=0.47). However, the post-hoc analysis demonstrated a consistent benefit with melflufen-dex in patients who have not received ASCT or progressed ≥36 months after receiving ASCT.68 The median OS was 23.6 months with melflufen-dex versus 19.8 months with pom-dex (HR: 0.83 [95% CI: 0.62–1.12]; p=0.22). Long-term results of the OCEAN study reported at 65th ASH Annual Meeting & Exposition (ASH 2023) were consistent with previous analyses demonstrating OS of 20.2 months versus 24.0 months (HR: 1.09 [95% CI: 0.88–1.35]; p=0.4088] in the ITT population and 23.6 months versus 19.1 months (HR: 0.88 [95% CI: 0.67–1.36]; p=0.3606) in patients without prior ASCT or progressed ≥36 months after ASCT with melflufen-dex versus pom-dex, respectively.69 Clinical benefits of melflufen-dex in the OCEAN study are summarized in Table 1. Remarkably, survival outcomes in del(17p) patient subgroup were consistent with the ITT population: the median PFS was 7.1 months versus 2.9 months (HR: 0.45 [95% CI: 0.26–0.79]; p=0.006), the median OS was 11.5 months versus 15.9 months (HR: 1.31 [95% CI: 0.76–2.26]; p=0.33) and ORR was 33.3% versus 10.8% (p=0.028) with melflufen-dex versus pom-dex, respectively.48,61
The most common grade 3–4 TEAEs with melflufen-dex included thrombocytopenia (78% vs 13% with pom-dex), neutropenia (64% vs 50% with pom-dex) and anemia (43% vs 19% with pom-dex).69 Serious AEs occurred in 43% of patients in the melflufen-dex arm versus 50% in the pomalidomide arm. TEAEs leading to dose reductions were reported in 52% versus 28% of patients and TEAEs leading to discontinuations in 30% versus 24% of patients with melflufen-dex versus pom-dex, respectively. The rates of fatal AEs were comparable in both groups (14% vs 15%). Health-related QoL with melflufen was maintained throughout the treatment in the OCEAN trial.71
ANCHOR
The open-label, multicenter, phase I/IIa ANCHOR trial evaluated melflufen-dex in combination with either daratumumab or bortezomib in patients with RRMM after 1–4 prior lines of therapy who were refractory to (or intolerant of) an IMiD and/or a PI.65 The study employed a 3+3 dose-escalation model to determine the MTD in phase I, followed by dose expansion in phase IIa to assess efficacy and safety. All patients received melflufen (30 mg or 40 mg IV on day 1 of each 28-day cycle) in combination with either daratumumab (16 mg/kg IV) and dexamethasone (40 mg PO weekly) or bortezomib (1.3 mg/m2 subcutaneously [SC] on days 1,4, 8 and 11) and dexamethasone (20 mg or 40 mg PO weekly). The primary endpoint in phase II was ORR, and the secondary endpoints included the best response, DoR, TTR, PFS, OS and safety. The ANCOR trial was prematurely closed due to a partial clinical hold issued on all melflufen trials by the U.S. FDA before all planned patients in the bortezomib arm were enrolled.
The combination of melflufen-dex with daratumumab showed an ORR of 73% (sCR, 3%; CR, 6%; VGPR, 24%) across 33 patients treated with melflufen doses of 30 mg and 40 mg.65 The median DoR was 12 months, the median PFS was 12.9 months and OS 26.1 months. In the melflufen-dex plus bortezomib arm (n=23), ORR was 78% (sCR, 4%; CR, 4%; VGPR, 22%), the median DoR was 15.8 months and the median PFS was 14.7 months. OS data were immature, with 17 (74%) patients still alive at the data cut-off.
The safety profile of both combinations were similar to that of melflufen-dex alone, with no dose-limiting toxicities reported with either melflufen dose.65 Most common any-grade and grade ≥3 TEAEs were cytopenias, which were clinically manageable with dose reductions and growth factor support. No dose-limiting toxicities were observed for either melflufen dose; in the melflufen-dex plus bortezomib arm, 30 mg melflufen was identified as the recommended dose for future studies.
LIGHTHOUSE
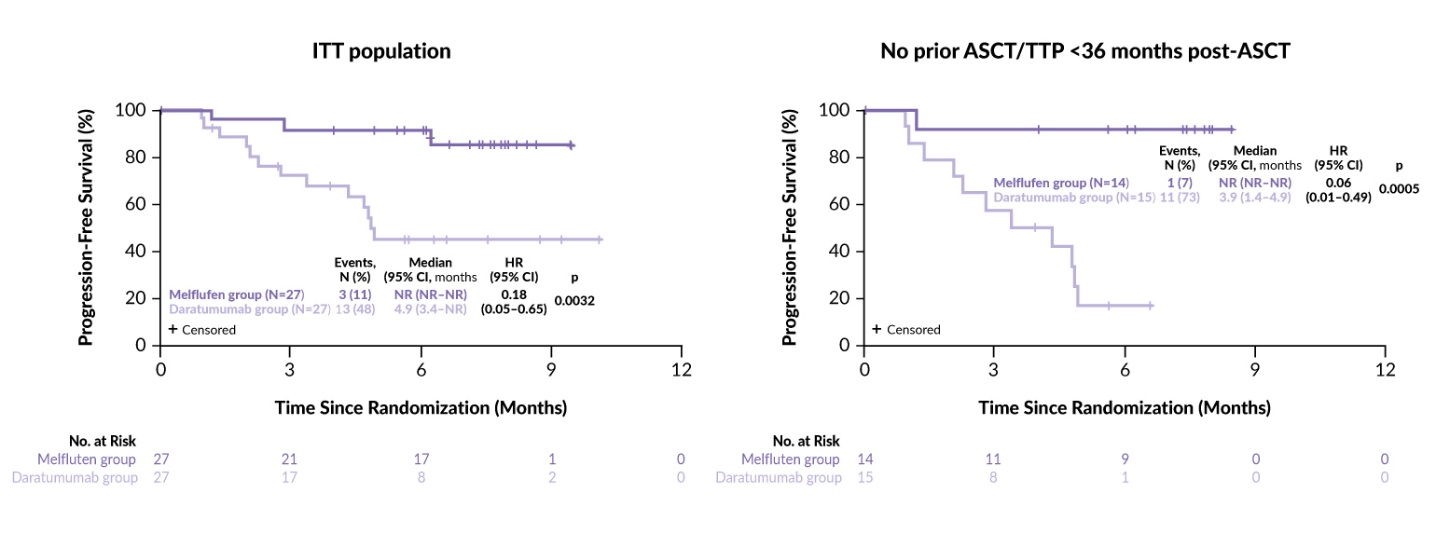
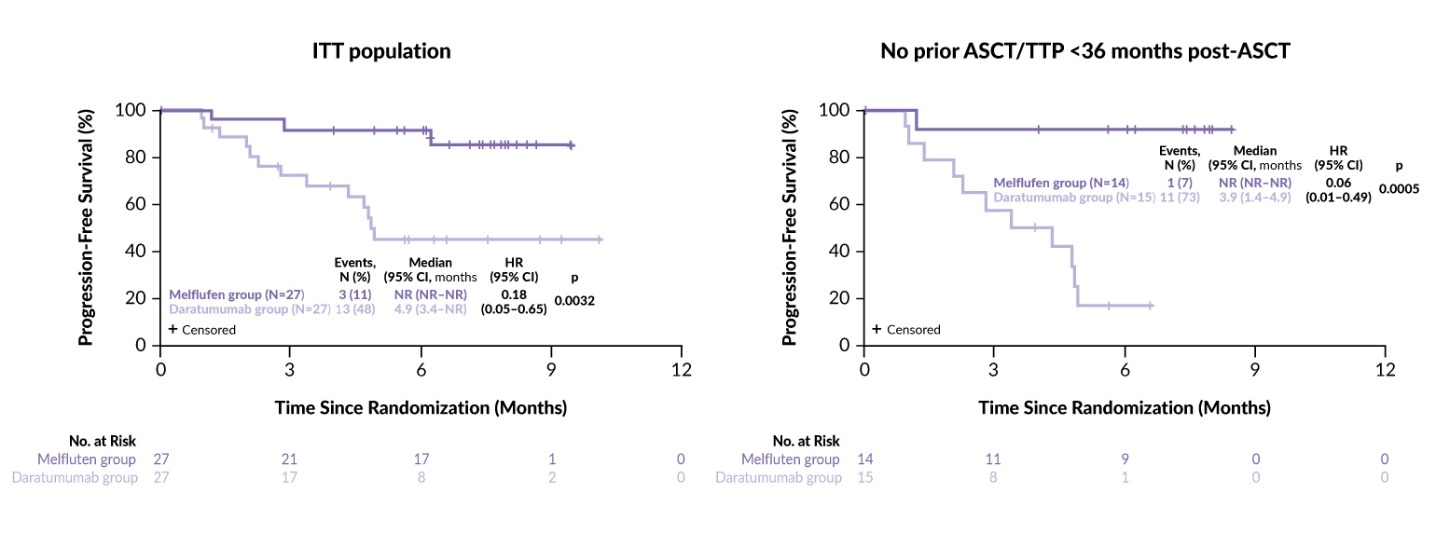
The randomized, phase III LIGHTHOUSE trial further assessed the combination of melflufen with daratumumab and dexamethasone versus daratumumab monotherapy in 240 patients with RRMM.66 Eligible patients (aged ≥18 years) were required to be refractory to both an IMiD and a PI or to have undergone at least three prior lines of therapy, including an IMiD and a PI. Patients were randomized 1:1 to receive either melflufen (30 mg IV on day 1 of each 28-day cycle) plus daratumumab (1800 mg SC) and dexamethasone (40 mg PO weekly) or daratumumab only. The treatment was continued until disease progression or unacceptable toxicity. The primary endpoint was PFS and the secondary endpoints were OS, ORR and safety. The trial included a crossover option to the melflufen-containing regimen for patients who experienced disease progression on daratumumab monotherapy. LIGHTHOUSE was closed prematurely after enrolling 54 patients (27 per arm) following the FDA directives based on a preliminary review of the OCEAN study.
The results showed a significant improvement in efficacy in the combination arm compared with daratumumab monotherapy. The median PFS was not reached in the melflufen arm versus 4.9 months in the daratumumab arm (HR: 0.18 [95% CI: 0.05–0.65]; p=0.0032) at a median follow-up time of 7.1 and 6.6 months, respectively (Figure 4). The ORR was 59% with the combination therapy versus 30% with daratumumab alone (p=0.0300). OS data were immature, with two events (7%) in the melflufen group and four events (15%) in the daratumumab group (HR: 0.47 [95% CI: 0.09–2.57]; p=0.3721). Efficacy endpoints were more prominently in favor of the melflufen combination among patients with no prior ASCT or TTP >36 months after a prior ASCT, with a median PFS not reached versus 3.9 months, respectively (HR: 0.06 [95% CI: 0.01–0.49]; p=0.0005) (Figure 4). The safety profile of the combination was consistent with that reported in the ANCHOR study.
Real-world data for melflufen in RMMM
A real-world study conducted at the Dana-Farber Cancer Institute (DFCI) evaluated the use of melflufen in combination with dexamethasone in 12 patients with RRMM (Dr Jacob Laubach and Dr Sharier Hussein, personal communication; now published in the European Journal of Hematology).72 Patients (median age, 74 years) had been diagnosed with MM a median of 11 years prior and had received a median of 5.5 lines of therapy. All patients had prior exposure to lenalidomide, bortezomib and daratumumab. Additionally, patients had been treated with carfilzomib (75%), pomalidomide (92%), cyclophosphamide (50%) and elotuzumab (25%). Five of the 12 patients had previously undergone ASCT. The analysis demonstrated an ORR of 55%, with three patients achieving CR and one achieving VGPR. Treatment discontinuations were primarily due to disease progression (58%), the market withdrawal of melflufen (25%), AEs (8%) and death in the context of progressive disease (8%). No cases of mucositis, alopecia or secondary malignancies were reported.
Regulatory approvals and market authorizations for melflufen
Melflufen was initially granted accelerated approval by the FDA in February 2021 in combination with dexamethasone for the treatment of adult RRMM patients who had received at least four prior lines of therapy, including a PI, an IMiD and anti-CD38 mAb. The approval was contingent upon confirmatory trials to verify its clinical benefit and was subsequently withdrawn in February 2024 based on the data from the OCEAN trial which did not demonstrate OS benefit in the ITT population as per the initial analysis, although further follow up has shown an improvement in hazard ratio to 1.09.73,74 In the European Union and the United Kingdom, melflufen has been fully approved in combination with dexamethasone for the treatment of adults with RRMM who have received at least three prior lines of therapies, whose disease is refractory to at least one PI, one IMiD and one anti-CD38 mAb, and who have demonstrated disease progression on or after the last therapy.75,76
As of April 01, 2025, the FDA lifted the clinical hold on OPD5, a next-generation PDC derivative of melflufen designed to offer a potentially improved risk/benefit profile along with enhanced intellectual property protection.77 A phase I OP-502 trial is in preparation to evaluate safety, tolerability and efficacy of OPD5.
Summary and future directions
Taken together, the clinical data demonstrate the benefits of melflufen in combination with dexamethasone as well as in regimens that include daratumumab or bortezomib. Melflufen plus dexamethasone has shown efficacy in heavily pretreated triple-class refractory patients, providing a therapeutic option for those with limited alternatives. The clinical benefits of melflufen are more pronounced in patients who have not undergone ASCT or who have experienced disease progression ≥36 months following ASCT, highlighting its potential in less alkylator-exposed populations. Furthermore, melflufen demonstrates activity in patients with high-risk cytogenetic features such as del(17p). Preclinical studies have revealed that melflufen retains efficacy in cell lines lacking p53 function, including those with p53 mutations, underscoring its ability to overcome the resistance mechanisms commonly associated with genetic aberrations. Melflufen has demonstrated a consistent safety profile throughout the clinical development program, primarily comprising hematological AEs that were predictable and manageable with dose modifications and supportive care. These findings position melflufen as a promising agent for addressing the unmet needs of RRMM patients.
Future strategies for melflufen research should focus on personalized treatments in RRMM, particularly targeting stemness characteristics and EMD. By increasing the susceptibility of myeloma cells to immune attacks, melflufen could synergize with mAbs and PIs, forming a backbone for combination regimens. Following the development of melflufen, new generations of PDCs are being designed to further enhance therapeutic outcomes in MM and other types of cancers by selectively targeting tumor cells with high peptidase activity. These advanced PDCs, such as OPDC3, exhibit improved stability, greater selectivity and enhanced cytotoxicity in preclinical models of MM, AML/MDS and diffuse large B-cell lymphoma, demonstrating up to 10-fold greater anti-MM activity over melflufen.78–80
Together with other emerging therapies, including CAR T-cell therapy, bispecific antibodies, CELMoDs, ADCs and targeted therapies, PDCs with alkylator payloads such as melflufen offer an important new option for improving outcomes in high-risk or heavily pretreated patient populations, with relatively simple out-patient based therapeutic translation to real world practice.81,82
Conflict of interest
Yuxin Liu, Shahrier Hossain, Taya Salman, Pedro Vianna and Jacob Laubach have no conflicts to disclose. Omar Nadeem reports honorarium from Pfizer; advisory board participation for BMS, Janssen, Takeda, Sanofi, GPCR Therapeutics; and research funding from Takeda, Janssen. Shonali Midha reported Abbvie stock ownership and honoraria for consultancy from Janssen and Pfizer. Monique Hartley-Brown received honoraria for consultancy from AbbVie, Bristol Myers Squibb/Celgene, GSK, Janssen, Karyopharm and Sanofi, and speaker’s honoraria from Multiple Myeloma Research Foundation and Cancer Care. Adam S. Sperling has acted as a consultant for Novartis and Roche. Giada Bianchi received honoraria for consultancy from Prothena. Omar Nadeem reports honorarium from Pfizer; advisory board participation for BMS, Janssen, Takeda, Sanofi, GPCR Therapeutics; and research funding from Takeda, Janssen. Clifton Mo acted on the advisory boards for AbbVie, BMS, GSK, Janssen, Karyopharm, Sanofi and Takeda, and as a consultant for AbbVie, Janssen, Karyopharm and Sanofi. Paul Richardson received honoraria for consultancy from Celgene/BMS, GSK, Karyopharm, Oncopeptides, Regeneron and Sanofi, and research grants from Oncopeptides.
Funding
The authors have declared that no financial support was received from any organization for the submitted work.
Author contributions
All authors contributed to and approved the final manuscript.