Introduction
Lung cancer is responsible for 18% of all cancer-related deaths worldwide and up to 19.5% in the European Union.1,2 Non-small cell lung cancer (NSCLC) is the most prevalent form of lung cancer, accounting for up to 85% of all cases.3 Despite the advent of new therapies, such as checkpoint inhibitors and targeted therapy options, the 5-year survival remains poor.4 Treatment of NSCLC poses many challenges due to tumor heterogeneity, the presence of diverse genetic mutations, resistance to therapy, late-stage detection, as well as multimorbidity, including the presence of chronic obstructive pulmonary disease (COPD), coronary heart disease and smoking history. These factors often limit treatment options, thus underscoring the need for personalized treatment approaches and ongoing research to improve outcomes for patients with NSCLC. Advancements in targeted therapies and immunotherapies have provided new avenues for managing the disease, improving overall survival and enhancing patient quality of life. Molecular findings in lung cancer evolve constantly, thereby creating space for the development of new targeted therapies for tumors with actionable genomic alterations.
The epidermal growth factor receptor (EGFR) functions as a receptor tyrosine kinase that regulates cell growth and division. Lung cancers with mutations in the EGFR gene account for approximately 19% of cases in the Western world and up to 50% of cases in the Asian population.5,6 The most common EGFR-activating mutations are deletions in exon 19 (Ex19del) and point L858R mutations in exon 21, which are detected in up to approximately 85% of newly diagnosed EGFR-positive NSCLC. Other alterations are categorized as uncommon EGFR mutations, which include exon 20 insertions (Ex20ins) accounting for approximately 10% of cases, as well as T790M, G719X, S768I and L861Q point mutations.7–9 Furthermore, the presence of two or more independent mutations in the EGFR gene is defined as compound mutations.
Bispecific antibodies harbor great potential in cancer therapy due to their innovative design and combined diverse mechanisms of action.10 By simultaneously binding to two different antigens or two epitopes of the same antigen, these engineered hybrid molecules can significantly enhance therapeutic efficacy by increasing anti-tumor response rates and redirecting immune cells to target tumor cells. The mechanisms of action of bispecific antibodies include dual inhibition of tumor signaling pathways, recruitment and activation of T cells to exert anti-tumor activity, immune checkpoint inhibition and forced formation of protein complexes on the cell surface. Several important updates on bispecific antibodies in NSCLC treatment were recently presented at the 2024 European Lung Cancer Congress (ELCC 2024) held in Prague, Czech Republic, on March 20−23, 2024.
Amivantamab is a bispecific antibody with the capability to activate immune cells that targets both the EGFR and mesenchymal epithelial transition (MET) receptor, the latter being a known mechanism of resistance to EGFR-targeting therapy.11–13 Amivantamab is approved for use as a single-agent therapy by Swissmedic and the European Medicinal Agency (EMA) and is administered every two weeks intravenously (IV) for treating patients with advanced NSCLC harboring EGFR Ex20ins mutations following the failure of platinum-based chemotherapy.14 A subcutaneous (SC) formulation of amivantamab has recently been developed and biweekly (Q2W) and triweekly (Q3W) doses have been previously evaluated.15,16 Data from the PALOMA study evaluating SC administration of a once-monthly amivantamab (Q4W) dose has recently been reported, demonstrating pharmacokinetic and safety profiles similar to its IV counterpart.17
The third-generation EGFR tyrosine kinase inhibitor (TKI) osimertinib is the current standard of care for treating EGFR-mutant NSCLC.18 The randomized, open-label, multicenter, phase III FLAURA trial showed an improved progression-free survival (PFS) in patients receiving osimertinib compared to standard first-generation TKIs (18.9 months vs 10.2 months) in EGFR-mutant (ex19del or L858R being the most frequent mutations) NSCLC.19 Regarding the median overall survival (OS), the benefit is also in favor of osimertinib (median OS, 38.6 months vs 31.8 months; HR: 0.80 [95.05% CI: 0.64–1.00]; p=0.046).20 With the wide use of osimertinib, resistance to this TKI occurs, as after a median time of 17.2 months, patients with common EGFR-mutant NSCLC face progression on osimertinib therapy sooner or later.21
Lazertinib, a highly effective, brain-penetrating third-generation EGFR TKI, in combination with amivantamab has demonstrated enhanced tumor growth suppression in treatment-naïve and osimertinib-relapsed EGFR-mutant, advanced NSCLC.18,22 Recent exploratory analyses from the phase III MARIPOSA trial have examined how interruptions in amivantamab dosing influence the effectiveness and safety of the initial treatment with amivantamab plus lazertinib for patients with advanced NSCLC harboring EGFR mutations.23 The latest findings suggest that amivantamab plus lazertinib sets a new benchmark for first-line treatment in patients with advanced EGFR-mutant NSCLC.
PALOMA: Novel dosing strategy for amivantamab in advanced solid tumors
PALOMA (NCT04606381) is an ongoing, open-label, phase Ib dose-escalation study of SC amivantamab in patients with advanced solid tumors who may benefit from EGFR- or MET-directed therapy.17 Of the 127 enrolled in PALOMA, 19 patients (median age, 62 years) received the SC Q4W dose of amivantamab. Notably, 17 (89%) of these patients had NSCLC.
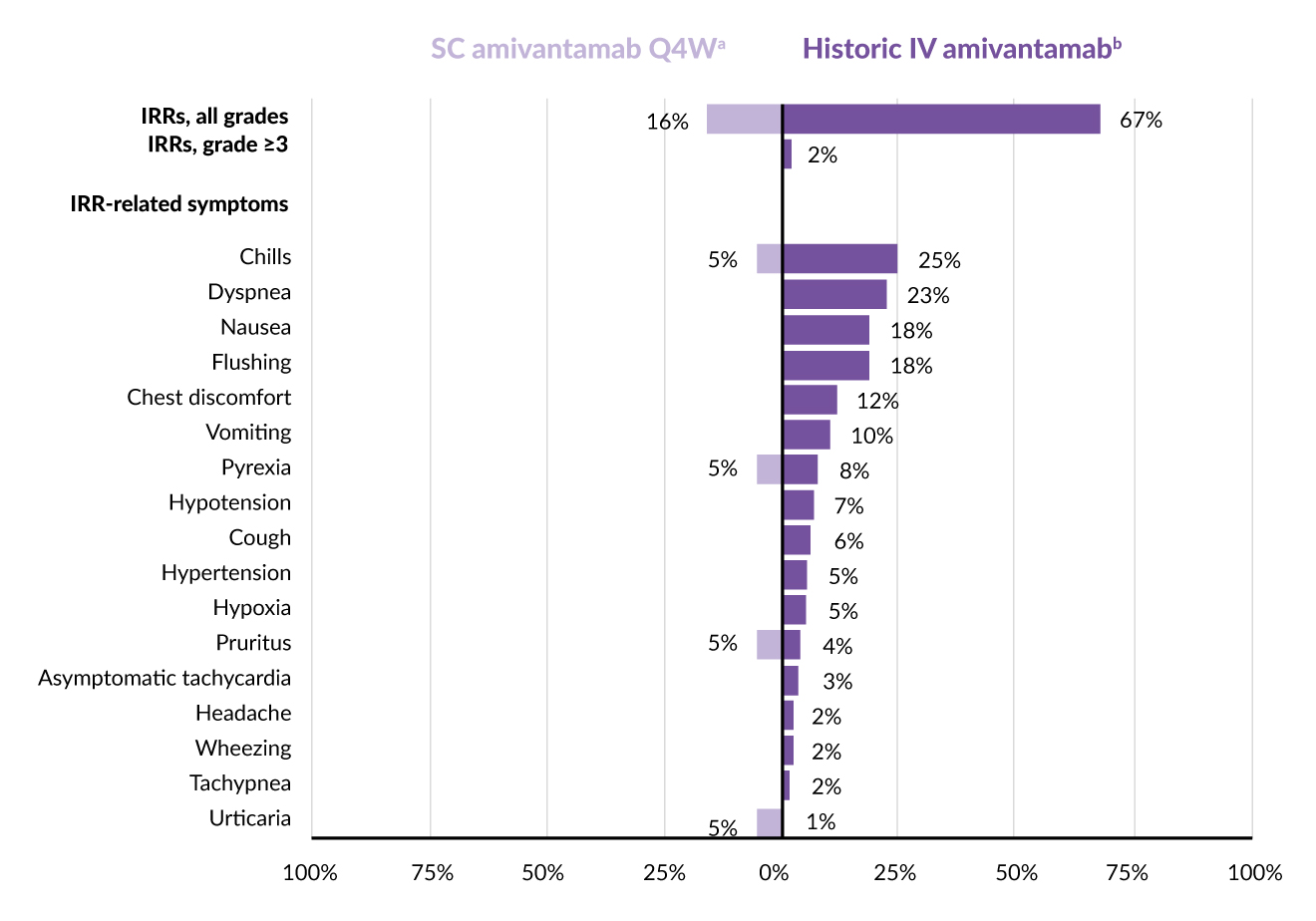
Three of the 19 patients (16%) reported infusion-related reactions (IRRs), all of which were mild to moderate (grade 1–2) and occurred after the first amivantamab SC dose (Figure 1).17 Symptoms associated with IRRs included fever, chills, itching and hives in one patient each. The most frequently observed adverse events (AEs) were skin rash (79%; mostly grade 1–2, with two instances of grade 3), nail infection (58%), muscle pain (47%), fatigue, nausea and mouth sores (32% each), followed by swelling of the limbs and fever (26% each). Eight patients (42%) had severe AEs of grade 3 or higher, with three cases (16%) being related to treatment (two instances of rash and one of low potassium level). Two patients stopped taking SC amivantamab due to AEs not related to the therapy.
Pharmacokinetic data showed that the administration time of SC amivantamab Q4W was between 7 and 10 minutes.17 The geometric mean values for the minimum concentration (Ctrough) and the area under the curve (AUC0−672h) during Cycle 2 for the evaluated SC Q4W dose were 325 μg/mL and 286,612 μg/h/mL, respectively. The Q4W dose was adjusted to 3,520 mg (or 4,640 mg for those weighing ≥80 kg) to align more closely with the steady-state Ctrough of the approved biweekly IV amivantamab dose. Simulated geometric mean ratios for the adjusted Q4W SC dose compared to the reference IV dose at steady state were 0.92 (90% CI: 0.76–1.11) for Ctrough and 1.27 (90% CI: 1.18–1.36) for AUC0−672h.
Overall, the once-monthly SC dose of amivantamab showed fewer IRRs and better tolerability than the intravenous form.17 This dosing strategy achieved exposure levels comparable to the approved IV dosage and is currently under further investigation in the phase II PALOMA-2 study (NCT05498428).24
_and_irr-related_symptoms_in_paloma.jpg)
MARIPOSA: Amivantamab plus lazertinib versus osimertinib in advanced NSCLC
MARIPOSA (NCT04487080) is a phase III, multicenter, randomized clinical trial designed to evaluate the efficacy and safety of combining amivantamab plus lazertinib compared to single-agent osimertinib in patients with untreated, advanced NSCLC that harbor EGFR mutations.25 Enrolling 1,074 patients, the MARIPOSA trial met its primary endpoint, showing a statistically significant and clinically meaningful improvement in PFS for patients treated with the amivantamab plus lazertinib combination compared to those receiving osimertinib (median PFS, 23.7 months vs 16.6 months; HR: 0.70 [95% CI: 0.58–0.85]; p<0.001).25,26 Notably, this was the first pivotal study to show a clinically meaningful benefit in a chemotherapy-free regimen versus osimertinib.25,26 Consistent and significant PFS benefits of amivantamab plus lazertinib versus osimertinib were also observed across all high-risk subgroups. These included patients with TP53 co-mutations (18.2 months vs 12.9 months; HR: 0.65 [95% CI: 0.48–0.87]; p=0.003), patients with detectable baseline EGFR-mutant circulating tumor DNA (ctDNA) by droplet digital polymerase chain reaction (ddPCR) (20.3 months vs 14.8 months; HR: 0.68 [95% CI: 0.53–0.86]; p=0.002), patients with a history of brain metastases (18.3 months vs 13 months; HR: 0.69 [95% CI: 0.53–0.92]; p=0.010) and those with liver metastases at baseline (18.2 months vs 11 months; HR: 0.58 [95% CI: 0.37–0.91]; p=0.017).27
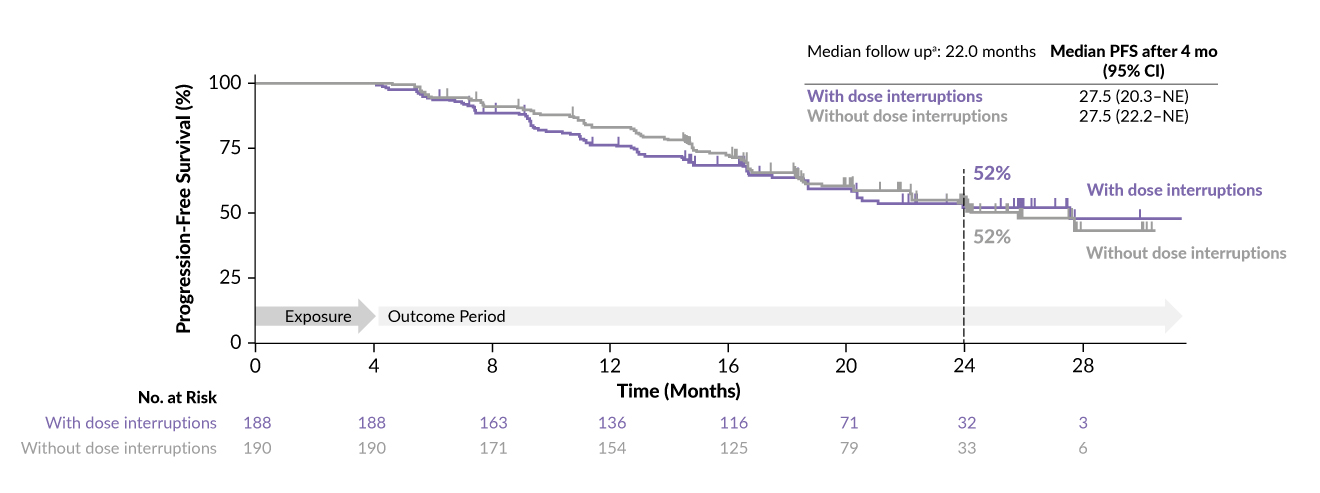
In a recent analysis focusing on patients assigned to the amivantamab plus lazertinib treatment group in the MARIPOSA study, 429 and 421 patients (median age, 62.5 years) were evaluated for efficacy and safety assessment, respectively.23 According to the study’s guidelines, amivantamab was to be adjusted in dosage before any modifications to lazertinib. Dose interruptions were identified as any pause in amivantamab treatment for any reason (skipping dose, dose reduction or drug discontinuation). Key findings revealed that out of the 421 patients who received at least one dose, nearly half (44.7%, or 188 patients) experienced amivantamab dose interruptions within the first four months of therapy. Of the 421 patients who received the combination, 43 were not included in this analysis for different reasons (study discontinuation, disease progression or death). Among the 188 patients with dose interruptions, the median time to first interruption was 43 days (interquartile range, 16–72). At a median follow-up period of 22 months, the median PFS for patients with early dose interruptions was 27.5 months (Figure 2). Baseline characteristics were similar between patients with and without dose interruptions. The analysis also showed that median PFS, objective response rate (ORR), and median duration of response (DoR) were comparable between patients with and without amivantamab dose interruptions in the initial four months and after the first four months, aligning with the overall results for the amivantamab plus lazertinib treatment arm.
AEs were primarily observed in the initial four months and showed a decline over the following four months.23 The incidence of rash decreased by approximately 50%, paronychia reduced by approximately 30% and diarrhea saw a significant drop of about 70%. Additionally, there were no reports of grade 4 or 5 AEs. The recommendation of dose interruption of amivantamab per protocol was for grade 2 or higher AE.
The conclusion drawn by the authors from this analysis is that early dose adjustments of amivantamab, as outlined in the MARIPOSA protocol, did not negatively affect the efficacy of the amivantamab plus lazertinib combination.23 These findings position amivantamab plus lazertinib as a new promising first-line treatment option for patients with EGFR-mutant advanced NSCLC, underscoring its effectiveness even with necessary dose modifications. Long-term follow-up and further data are needed to determine the optimal dosage for managing toxicity associated with this regimen, as well as to identify patient subgroups who would benefit most from this regimen and in whom it can replace single-agent osimertinib.
_after_4-month_amiva.jpg)
Conclusions
Data from the PALOMA study demonstrated that a once-monthly SC dose of amivantamab showed fewer IRRs and better tolerability than the intravenous form. The MARIPOSA study positioned the combination of amivantamab and lazertinib as a new promising first-line treatment option for patients with EGFR-mutant advanced NSCLC, demonstrating its effectiveness even with necessary dose modifications.
Conflict of interest
Prof. Alfredo Addeo received honoraria for consulting or advisory role from Bristol-Myers Squibb, AstraZeneca, Roche, Merck Sharp & Dohme, Pfizer, Eli Lilly, Astellas, Amgen, Novartis and Merck, served on the speaker bureau of Eli Lilly, AstraZeneca, and Amgen and received a grant from AstraZeneca. These funding entities did not play a role in the development of the manuscript and did not influence its content in any way. Other authors have declared that the manuscript was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors have declared that no financial support was received from any organization for the submitted work.
Author contributions
All authors contributed to and approved the final manuscript.